
Treacher Collins sendromu kısa adıyla TCS, kafa ve yüzdeki belirgin anormallikler ile karakterize nadir bir genetik hastalıktır. Baş ve yüzde görülen anormallikler, solunum ve beslenme zorluğuna neden olabilen zigomatik kompleksin yani elmacık kemiklerinin, çenenin, damak ve ağız boşluğunun gelişmemiş kalmasını içerir. Ayrıca, etkilenen bireyler üst ve alt göz kapakları arasındaki açıklığın aşağı doğru bir eğimine ve işitme kaybına neden olan dış ve orta kulak yapılarının kusurlarına sahiptir. Mikrosefali ve psikomotor gecikmesi gibi beyin ve davranış anomalileri de zaman zaman durumun bir parçası olarak rapor edilmiştir.
TCS ile ilişkili spesifik semptomlar ve fiziksel özellikler, bir kişiden diğerine büyük ölçüde değişebilir. Bazı bireyler tanı konamayacak kadar hafif derecede etkilenebilirken bazıları ciddi, hayatı tehdit edici komplikasyonlar geliştirebilir. TCS’deki TCOF1, POLR1C, veya POLR1D genlerinin bir mutasyonudur. TCOF1 durumunda kalıtım şekli otozomal baskın iken POLR1C için otozomal çekimserdir. Buna karşılık, POLR1D’de hem otozomal baskın hem de çekimser mutasyonlar, TCS ile birlikte rapor edilmiştir. TCS, ilk kez 1900 yılında tıp literatüründeki bozukluğu tanımlayan Londra oftalmolog Edward Treacher Collins’in adını almıştır. Ayrıca mandibulofasiyal distostoz veya Treacher Collins-Franceschetti sendromu olarak da bilinir.
Belirtileri
TCS’nin semptomları ve ciddiyeti, aynı ailenin üyeleri arasında bile bir kişiden diğerine büyük ölçüde değişebilir. Bazı bireyler, teşhisine kadar fark edilemeyecek düzeyde hafif etkilenebilirler. Bazıları ise önemli anormallikler ve hayatı tehdit edici solunum sistemi komplikasyonları yaşabilirler. Etkilenen bireylerin aşağıda belirtilen tüm belirtilere sahip olmayacağını not etmek önemlidir.
TCS’nin ana karakteristik özellikleri yüzün belli kemiklerini, kulakları ve göz çevresindeki yumuşak dokuları kapsar. Etkilenen bireyler belirgin yüz özelliklerine sahiptir ve potansiyel olarak işitme ve görme sorunları geliştirir. TCS anormallikleri, tipik olarak simetriktir (yüzün her iki tarafında neredeyse aynıdır) ve doğuştandır. Konuşma ve dil gelişimi, işitme kaybı, yarık damak veya çene ve hava yolu problemleriyle tehlikeye girebilir. Zekâ; genellikle etkilenmez, ancak mikrosefali ve bilişsel gecikme gibi beyin ve davranış anomalileri durumun bir parçası olarak nadiren bildirilmiştir.
TCS’li bebekler az gelişmiş veya eksik elmacık kemikleri sergiler, bu da 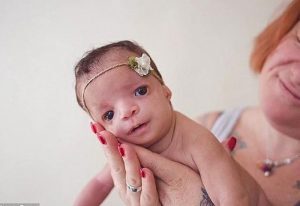 yüzün bu alanının düz veya çökük görünmesine neden olur. Alt çenekemiği olan mandibula, tam olarak gelişmemiştir ve çenenin ve alt çenenin anormal derecede küçük görünmesine neden olur. Alt çene kemiğinin bazı kısımlarını kaslara bağlayan bazı kemik yapıları alışılmadık şekilde düz veya yok olabilir. Etkilenen bebekler de boğazda azgelişmişlik gösterebilirler. Alt çenenin az gelişmiş olması veya çenenin anormal küçüklüğünde farengeal hipoplazisi erken bebeklik döneminde beslenme problemlerine veya solunum yetmezliğine katkıda bulunabilir. Çocuklar, normal solunum ve uyku sırasında hava hareketinin kısa süreli tekrarlanmasıyla karakterize obstrüktif uyku apnesi yaşayabilir. Ağır derecede etkilenen bazı kişilerde hayatı tehdit eden solunum güçlüğü gelişebilir.
yüzün bu alanının düz veya çökük görünmesine neden olur. Alt çenekemiği olan mandibula, tam olarak gelişmemiştir ve çenenin ve alt çenenin anormal derecede küçük görünmesine neden olur. Alt çene kemiğinin bazı kısımlarını kaslara bağlayan bazı kemik yapıları alışılmadık şekilde düz veya yok olabilir. Etkilenen bebekler de boğazda azgelişmişlik gösterebilirler. Alt çenenin az gelişmiş olması veya çenenin anormal küçüklüğünde farengeal hipoplazisi erken bebeklik döneminde beslenme problemlerine veya solunum yetmezliğine katkıda bulunabilir. Çocuklar, normal solunum ve uyku sırasında hava hareketinin kısa süreli tekrarlanmasıyla karakterize obstrüktif uyku apnesi yaşayabilir. Ağır derecede etkilenen bazı kişilerde hayatı tehdit eden solunum güçlüğü gelişebilir.
Solunum veya beslenme güçlüğüne katkıda bulunabilecek ek anormallikler, nazal hava yollarının daralmasını veya tıkanmasını içerir. Bazı çocuklar, ağız çatısı tamamen kapanmamış ağızdan daha uzağa kaydırılmış bir dille tanımlanan, şiddetli mikrognatiyi içeren “Pierre Robin Dizisi” nin özelliklerine sahip olarak tanımlanabilir. Ek olarak, ağız ve çenedeki kusurlar, az gelişmiş veya yanlış hizalanmış dişler gibi diş anormallikleri ile sonuçlanabilir. Ayrıca, eksik dişler, diş minesinin bulanıklaşması veya renk atması gibi ek diş anormallikleri de bildirilmiştir. Ve bazı üst dişleri uyumsuz çıkan TCS’li bireyler, orta kulaktan iletilecek ses dalgalarının başarısızlığından dolayı işitme kaybı gelişebilir. İletken işitme kaybı genellikle orta kulak içindeki yapıları etkileyen anormalliklerden kaynaklanır ve bireylerde ayrıca ses dalgalarının orta kulakta iletildiği üç küçük kemik de bozuk veya eksik olabilir. Ayrıca dış kulak yapıları, dış kulak kanallarının daralması veya tıkanması ile küçük veya bozuktur ve ya sıklıkla bulunmaz. Dış kulaklar buruşmuş veya döndürülmüş olabilir. Buna karşılık; kemik sarmal organının iç kulaktaki ve iç kulağın içindeki dengede bir rol oynayan yapılarında deformasyon olmasına rağmen iç kulak genellikle etkilenmez. Ek semptomlar, dış kulağın hemen önündeki cilt veya çukur oluşumlarının varlığını ve kulakların buruna doğru sarkan anormal bir geçişi içerebilir.
TCS’li birçok bebek, gözleri çevreleyen dokuda anormallikler gösterir. Bu göz farklılıkları, etkilenen bireylere üzülmüş bir yüz görünümü verebilir. En sık görülen oküler semptom, üst ve alt göz kapakları arasındaki açıklığın aşağıya doğru sarkmaya meyilli olmasıdır. Ayrıca alt göz kapağı çentiği ya da eksik kapak dokusunun yarıklığı, alt göz kapağı üzerinde kirpiklerin kısmen bulunmaması, şaşılık ve daralmış gözyaşı kanalları bulunur. Nadiren kusurlar gözle görülür ve irisin eksik dokusundaki çentik, yarık veya anormal derecede küçük gözler içerebilir. Bazı hastalarda görme kaybı olabilir. Görme bozukluğunun derecesi, oküler anormalliklerin ciddiyetine ve kombinasyonuna bağlı olarak değişir. Düşük göz kapağı anormallikleri gözlerin kurumasına neden olabilir.
TCS’li bazı kişiler, göz aralığındaki açıklık, üst göz kapağındaki çentik, burun deformitesi, anormal derecede geniş bir ağız, çok kavisli damak, kafadaki saçlı derinin yanaklara doğru uzaması yada olağandışı sarkması gibi ek fiziksel anormallikler sergiler. TCS’li bireylerin yaklaşık % 5’inde gelişim eksikliği ve psikomotor gecikme gibi nörolojik problemler görülür. Ancak, genellikle dil gelişimi etkilenmez. Bununla birlikte, işitme kaybı, yarık damak veya yapısal bozulma nedeniyle ses üretme güçlüğü nedeniyle konuşma gelişimi ile ilgili sorunlar ortaya çıkabilir.
Nedenleri
TCS, TCOF1, POLR1 C veya POLR1D genlerinin mutasyonundan kaynaklanır. TCOF1 durumunda kalıtım şekli otozomal baskındır, POLR1C için otozomal çekimserdir ve POLR1D için otozomal baskın veya otozomal çekimser olabilir.
Genetik hastalıklar, baba ve anneden alınan kromozomlar üzerindeki belirli bir özellik için genlerin kombinasyonu ile belirlenir. Baskın genetik bozukluklar, hastalığın ortaya çıkması için anormal bir genin sadece bir kopyasının gerekli olduğu durumlarda ortaya çıkar. TCS için TCOF1 ve POLR1D anormal gen, her iki ebeveynden kalıtsal olabilir veya etkilenen bireyde yeni bir mutasyonun sonucu olabilir. TCS’li vakaların yaklaşık % 60’ında, mutasyon bozukluğunun sebebi, önceki bir aile öyküsü olmadan rastgele gerçekleşen yeni bir mutasyondur. Bununla birlikte, bir ebeveyn hafifçe etkilenebilir ve bu hastalığa sahip olduklarının farkında olmayabilir. Anormal genin etkilenen ebeveynden çocuklarına geçme riski, önceki gebeliklerin etkilenmiş veya etkilenmemiş olduğuna bakılmaksızın her hamilelik için % 50’dir.
Resesif genetik bozukluklar bir birey her bir ebeveynden aynı özellik için aynı anormal geni miras aldığında meydana gelir. Bir birey hastalık için bir normal gen ve bir hasta gen alırsa kişi hastalık için taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte % 25’tir. Ebeveynler gibi taşıyıcı olan bir çocuğa sahip olma riski her hamilelikte% 50’dir. Bir çocuğun her iki ebeveynden normal gen alma ve bu özellik için genetik olarak etkilenmemiş olma şansı % 25’tir.
Araştırmacılara göre, TCOF1’in mutasyonlarının Kromozom 5’in uzun kolunda bulunan gen, çoğu TCS vakasına neden olur. Canlı hücrelerinin merkezinde bulunan kromozomlar her canlının genetik
 Kodlarını taşır. İnsan vücudu hücrelerinin normalde 46 kromozomu vardır. İnsan kromozomlarının çiftleri 1 ila 22 arasında numaralandırılmıştır ve cinsiyet kromozomları X ve Y olarak belirtilmiştir. Erkeklerde bir X ve bir Y kromozomu vardır ve dişilerde iki X kromozomu vardır. Her kromozomun “p” işaretli kısa bir kolu ve “q” işaretli uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok gruba bölünmüştür. Örneğin, “kromozom 5q32”, kromozom 5’in uzun kolundaki bant 32’yi belirtir. Numaralı bantlar, her bir kromozomda bulunan binlerce genin konumunu belirtir.
Kodlarını taşır. İnsan vücudu hücrelerinin normalde 46 kromozomu vardır. İnsan kromozomlarının çiftleri 1 ila 22 arasında numaralandırılmıştır ve cinsiyet kromozomları X ve Y olarak belirtilmiştir. Erkeklerde bir X ve bir Y kromozomu vardır ve dişilerde iki X kromozomu vardır. Her kromozomun “p” işaretli kısa bir kolu ve “q” işaretli uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok gruba bölünmüştür. Örneğin, “kromozom 5q32”, kromozom 5’in uzun kolundaki bant 32’yi belirtir. Numaralı bantlar, her bir kromozomda bulunan binlerce genin konumunu belirtir.
TCOF1, ihanet olarak bilinen bir proteini kodlayan (yaratan) talimatlar taşır. İhanetin TCS gelişiminde oynadığı kesin rol tam olarak anlaşılmamıştır. Araştırmacılar, melasın proteinleri (ribozomlar) birleştiren hücrelerde bulunan bazı küçük yapıların oluşturulmasında rol oynadığını belirlediler. Bu, embriyonik gelişim sırasında çok erken meydana gelen ve yüzün altındaki kemik ve kıkırdakların çoğuna yol açan nöral kret hücreleri adı verilen bir hücre grubunun oluşması için özellikle önemlidir. Ribozomların oluşumundaki kusurlardan kaynaklanan durumlar ribozomopatlar olarak adlandırılır. POLR1C ve POLR1D, RNA polimeraz I ve III’ün alt birimlerini kodlayan ribozom biyojenezi için de gereklidir. Mutasyonların muhtemel olduğu anlaşılıyor. TCOF1, POLR1C ve POLR1D nedeni yetersiz protein montaj ve hücreler embriyonun gelişimi sırasında nöral krest hücrelerinin çoğalma ve büyüme ihtiyaçlarını karşılamak için izin vermez. TCS oldukça değişken olduğundan, araştırmacılar ek genetik ve muhtemelen çevresel faktörlerin hastalığın değişken ciddiyetinde de rol oynayabileceğini düşünmektedir. Bu kavramı desteklemek için, son deneysel veriler, melanın nöral hücrelerde oksidatif stresin yol açtığı DNA hasarına karşı ve ayrıca her ikisinin de baş ve yüz gelişimini etkileyen nöral hücre bölünmesi sırasında iğ oryantasyonunda korunmada kritik bir rol oynadığını göstermiştir.
TCS’nin özellikleri ve fiziksel bulgularına sahip bazı bireyler (yaklaşık % 10-15), yukarıda belirtilen üç genin hiçbirinde mutasyona sahip değildir. Bu, henüz tanımlanmamış genlere ilave olarak bazı durumlarda TCS’ye neden olabileceğini düşündürmektedir.
Etkilenen Nüfuslar
TCS erkekleri ve kadınları eşit sayıda etkiler. Genel popülasyonda prevalansın 10.000-50.000 birey arasında 1 olduğu tahmin edilmektedir. Hafif derecede etkilenen bazı kişiler teşhis edilmeyebilir, bu da hastalığın genel popülasyondaki gerçek sıklığını belirlemeyi zorlaştırır.
Teşhis
Tam bir klinik değerlendirmeye, ayrıntılı bir hasta geçmişine ve karakteristik fiziksel bulguların tanımlanmasına dayanarak TCS tanısı konulur. Dış kulağın kusurları veya yokluğu gibi birçok anormallik doğumsaldır.
Klinik Test ve Çalışmalar
Özel X-ışını çalışmaları, gözlenen bazı kraniyofasiyal anormalliklerin varlığını veya kapsamını doğrulayacaktır. Örneğin, bu tür görüntüleme testleri, alt çene kemiğinin yeterince gelişmemiş olması, kafatasının belirli kısımlarını etkileyen hipoplazinin varlığı ve kapsamı, kafatasının varlığı nedeniyle çenenin anormal küçüklüğünü gösterir. Klinik değerlendirme sırasında görülemeyen kulağın ek kusurları mevcut olabilir. Ek olarak, az etkilenen bireylerde, ayrıntılı bir klinik inceleme ve kraniyofasiyal bölgenin X-ışını görüntülemesi, TCS ile ilişkili belirli karakteristik hafif varlığını gösterebilir. TCS diğer kraniyofasiyal sendromlarda oluşabilecek bazı fiziksel özellikleri paylaştığı için, birçok araştırmacı, moleküler genetik testler ve bazı durumlarda dikkatli, ayrıntılı bir aile öyküsü ile tanısal teyit yapılmasını önermektedir.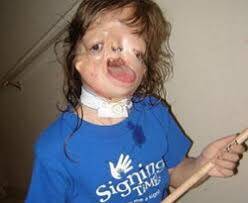
Teşhisi doğrulamak için moleküler genetik testler, TCOF1, POLR1C, ve POLR1D genlerindeki mutasyonları tespit etmek için ticari ve akademik araştırma laboratuvarları aracılığıyla yapılabilir. Bireylerin yaklaşık% 90-95’i TCOF1 geninin tanımlanabilir bir mutasyonuna sahiptir. Ayrıca, bir TCOF1, POLR1C veya POLR1D’nin genetik doğrulaması; mutasyon, doğumdan önce etkilenen bir ailede bir tanımlanmışsa, amniyosentez ve koryon villus örneklemesi ile tespit edilebilir. Bazı durumlarda, gelişen fetüsün bir görüntüsünü oluşturmak için yansıtılmış ses dalgalarını kullanan fetal ultrasonografi, TCS’yi düşündüren karakteristik bulguları ortaya çıkarabilir. TCS teşhisi konan bireyin akrabaları, özellikle ebeveynleri ve kardeşleri, hafif vakalar sıklıkla tanınmayacak ve teşhis edilmediği için dikkatlice incelenmelidir.
Tedaviler
TCS’nin tedavisi yoktur. Tedavi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını gerektirebilir. Pediatristler, pediatrik kulak, burun ve boğaz uzmanları, pediatrik diş hekimi, pediatrik hemşire, plastik cerrah, konuşma patologları, odyologlar, göz doktorları, psikologlar, genetikçiler ve diğer sağlık profesyonellerinin, bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilir.
Doktorlar, hastalıkla ilişkili olabilecek bazı anormallikleri tespit etmek için TCS’li bireyleri düzenli olarak izler. Örneğin, etkilenen bir kişinin işitme duyusu, işitme kaybının başladığını tespit etmek için dikkatle izlenmelidir. Bir bebeğin işitme duyusunun değerlendirilmesi çok önemlidir. Tam bir değerlendirme, doğru konuşma gelişimini sağlamak için yaşamın erken döneminde, hatta bir yaşından önce ve sonrada yıllık olarak yapılmalıdır.
Herhangi bir görme bozukluğu olasılığını tespit etmek için gözün içini görselleştirmek için oftalmoskop adında bir araç kullanılır. Bu inceleme, 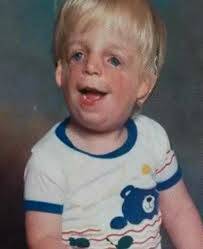 TCS ile bağlantılı olarak gözlerde anormallikler sergileyen hastalar için uygun önleyici adımlar veya acil tedaviyi sağlamak için önemlidir. Etkilenen bireyler de çene ve diş anormallikleri açısından izlenmelidir. Erken müdahale, etkilenen çocukların potansiyellerine ulaşmalarını sağlamak için önemlidir. Yararlı olabilecek özel hizmetler konuşma terapisi, özel sosyal destek ve diğer tıbbi, sosyal ve mesleki hizmetleri içerebilir. Bu sendorumundan mustarip olan aile ve bireyler uzaman genetik bir danışmandan yardım alabilirler.
TCS ile bağlantılı olarak gözlerde anormallikler sergileyen hastalar için uygun önleyici adımlar veya acil tedaviyi sağlamak için önemlidir. Etkilenen bireyler de çene ve diş anormallikleri açısından izlenmelidir. Erken müdahale, etkilenen çocukların potansiyellerine ulaşmalarını sağlamak için önemlidir. Yararlı olabilecek özel hizmetler konuşma terapisi, özel sosyal destek ve diğer tıbbi, sosyal ve mesleki hizmetleri içerebilir. Bu sendorumundan mustarip olan aile ve bireyler uzaman genetik bir danışmandan yardım alabilirler.
Cerrahi Müdahale
Bazı hastalarda, kraniyofasiyal kusurları, cerrahi müdahaleye gerek duyabilir. Damak yarığı onarımı, çeneyi yeniden yapılandırmak veya kafatasındaki diğer kemikleri onarmak için cerrahi yapılabilir. Kullanılan spesifik cerrahi prosedürler ve ameliyatın yapıldığı yaş, kusurların ciddiyetine, genel sağlık durumuna ve kişisel tercihlere bağlıdır. Örneğin, farklı anormallikler farklı yaşlarda tedavi edilebilir. Yarık damak genellikle 1-2 yaş civarında düzeltilir. Zigomatik ve yörünge rekonstrüksiyonu genellikle 5-7 yaş civarında meydana gelir. Dış ve iç kulak rekonstrüksiyonu genellikle 6 yaş civarında meydana gelir. Çene kemiğinin uzatılması veya yeniden yapılması, durumun derecesine ve ciddiyetine bağlı olarak yenidoğan ile genç yaşlar arasında değişebilir.
Obstrüktif hava yolları, ebeveynler veya klinisyenler için her zaman açık olmayan ciddi bir problem olabilir. Engelin ciddiyetini belirlemeye yardımcı olmak için bir uyku veya şekerleme çalışması kullanılabilir ve tedavi planını etkileyebilir. Şiddetli etkilenen bireylerde, trakeostomi adı verilen bir prosedür olan etkili bir havayolunu korumak için ön boruya yani trake borusuna cerrahi olarak yerleştirilebilir. Çene kemiğinin uzunluğunu arttırmak için kullanılan mandibular distraksiyon olarak bilinen bir prosedür gerekebilir. Beslenme güçlüğü çeken bebeklerin yeterli miktarda kalori almasını sağlamak için mideye bir tüp cerrahi olarak yerleştirilebilir. TCS ile potansiyel olarak ilişkili olan çeşitli kranyofasiyal anormallikleri tedavi etmek için çoklu ameliyatlar gerekebilir. Ameliyat sayısına rağmen, sonuçlar bir kişiden diğerine değişiklik gösterir ve sonuç nadiren tamamen düzelticidir.
Bazı kişilerde, orta kulak kusurlarının düzeltilmesi ve ilgili iletken işitme kaybının düzeltilmesi için bir işlem yapılabilir. Ancak, kemik bağlantılı işitme cihazları gibi özel işitme cihazları çoğu hastada ameliyattan ziyade yeterli olabilir. Kemik ankrajlı işitme cihazları, dış kulak kanalını ve orta kulağı etkileyebilir. İşlevsel olarak dış kulak kusurlarının düzeltilmesine yardımcı olmak için rekonstrüktif cerrahi uygulanabilir. Kozmetik sebeplerle, genellikle dış kulak rekonstrüksiyonu önce yapılmalıdır.
Ek Tedaviler
Göz anormallikleri ve buna bağlı görme bozukluğu gösteren TCS’li bireylerde, bazı durumlarda görüşün iyileştirilmesine yardımcı olmak için düzeltici gözlük, kontakt lensler, cerrahi ve diğer destekleyici teknikler kullanılabilir. Diş anormalliklerini düzeltmek için yapay, diş implantları, diş telleri, diş cerrahisi gibi diğer düzeltici prosedürler kullanılabilir.
Anestezi İle İlgili Önemli Noktalar
TCS ile ilişkili yapısal hava yolu problemleri, anestezistlerin ameliyat sırasında bir hava yolunu yönetmelerini ve bakımını yapmalarını zorlaştırabilir. Anestezi stratejisini en iyi şekilde planlamak için kapsamlı bir preoperatif değerlendirme ve tam bir klinik öykü içeren doğru değerlendirme yapılmalıdır.
Araştırma Terapileri
Tcof1, Polr1c ve Polr1d TCS’nin hayvan modelleri, vücudun zarar görmüş, hasta veya istenmeyen hücreleri öldürmesine yardımcı olan p53 proteinini anormal şekilde aktive olur. Böylelikle kranial sinir kret hücrelerinin kaybına ve dolayısıyla kraniyofasiyal kemik ve kıkırdak TCS’nin karakteristik özelliklerine neden olur. Araştırmacılar, p53 fonksiyonunu inhibe etmenin veya p53 aktivasyonuna yol açan mekanizmaları, TCS’nin gelişmesini önlemek için mümkün olan terapötik tedaviler olarak bloke etmenin yollarını araştırıyorlar. İnsanlarda TCS’yi taklit eden hayvan modelleri çalışmalarındaki son gelişmeler, diyet antioksidan takviyesinin,  embriyogenez sırasında sinir hücrelerini hasara karşı koruyabileceğini ve normal kraniyofasiyal gelişimi kolaylaştırabileceğini göstermektedir.
embriyogenez sırasında sinir hücrelerini hasara karşı koruyabileceğini ve normal kraniyofasiyal gelişimi kolaylaştırabileceğini göstermektedir.
Bazı araştırmacılar, TCS gibi kranyofasiyal bozukluğu olan bireylerde, cerrahi sonuçların iyileştirilmesi için ek bir tedavi olarak yağ dokusunda bulunan sap hücrelerinin kullanımını araştırmaktadır. İlk sonuçlar, etkilenen alanların büyümesini teşvik etmek için bu kök hücreler kullanılarak cerrahi sonuçların geliştirilebileceğini göstermiştir. Bununla birlikte bu terapi, deneysel ve tartışmalıdır ve potansiyel bir terapi olarak uygulanabilirliğini belirlemek için daha fazla araştırma gerektirir.
Kaynakça:
ghr.nlm.nih.gov
seattlechildrens.org
www.webmd.com
Yazar: Özlem Güvenç Ağaoğlu