
Konjenital aural atrezi, farklı şiddette olabilen, sadece dış kulağı veya orta kulağı da etkileyebilen bir kulak bozukluğudur. Kulak malformasyonu tek bir problem olabilir veya sendromik bir tablonun parçası olabilir. CCA’dan birinci ve ikinci branşiyal arkların ve birinci brankial yarıkların değişmiş gelişimi sorumlu olabilir. Schuknecht yüksek çözünürlüklü bilgisayarlı tomografi (BT) taraması ve cerrahi bulguların kombinasyonuna dayanarak dört derece ciddiyeti sınıflandırılmaktadır
• Tip A: fibrokartilajinöz kanalın daralması ve stenotik bölgenin distalinde kolesteatom varlığı
• Tip B: Kanalın fibrokartilajinöz ve kemikli kısmının daralması ve kıvrımlılığı, genellikle anormal timpanik membran ve malleus ile ilişkilidir.
• Tip C: Farklı fibröz doku ve kemik kombinasyonu ile tam atrezi. Tipik olarak, malleus ve incus birbirine kaynaşır, manubrium ve timpanik membran eksiktir ve stapes hareketlidir
• Tip D: Mastoidin pnömatizasyonunda azalma olan tamamen atrezi. Tip C’den daha şiddetli kemikçik anomalileri ve sıklıkla yüz siniri anormaldir
Başka araştırmacılar da CAA’yı sınıflandırmışlardır ve en ünlülerinden biri, bu malformasyonları üç tipte (A, B ve C) sınıflandıran Weerda’dır. Altmann ise üç kategoriyi tanımlayarak CAA’nın şiddetini ilişkilendiren histopatolojik bir sınıflandırma tanımlamıştır: hafif, orta ve ciddi şekilde hatalı biçimlendirilmiş türler. Birçok yazar o zamandan beri bu sınıflandırma sistemini değiştirmiş, cerrahi bulgular ve fonksiyonel sonuca dayalı olarak tip II’yi daha da alt sınıflandırmıştır.
CAA’nın Embriyolojisi ve Etiyopatogenezi
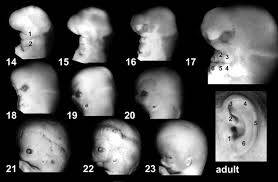 Araştırmacılara göre, aurikular gelişime katkıda bulunur ve her ikisi de CAA’nın etiyopatogenezinde rol oynayabilir. Kulak kepçesi, embriyonik yaşamın üçüncü ila altıncı haftaları arasında, kemerler üzerinde tepecikler belirdiğinde gelişmeye başlar ve oluşumu gebeliğin dördüncü ayında tamamlanır. Tragusun temeli, sarmal kök ve sarmalın üst kısmı, ilk kemerden türetilen ön üç tepeden gelir. İkinci arktan çıkan arka tepecik, antiheliks, antitragus ve lobül oluşumundan sorumludur.
Araştırmacılara göre, aurikular gelişime katkıda bulunur ve her ikisi de CAA’nın etiyopatogenezinde rol oynayabilir. Kulak kepçesi, embriyonik yaşamın üçüncü ila altıncı haftaları arasında, kemerler üzerinde tepecikler belirdiğinde gelişmeye başlar ve oluşumu gebeliğin dördüncü ayında tamamlanır. Tragusun temeli, sarmal kök ve sarmalın üst kısmı, ilk kemerden türetilen ön üç tepeden gelir. İkinci arktan çıkan arka tepecik, antiheliks, antitragus ve lobül oluşumundan sorumludur.
Orta kulak boşluğu gebeliğin 4. haftasından itibaren ilk faringeal arktan kaynaklanır. Pinna, embriyolojik yaşamın 28. haftasında kanalize olan dış etin çevresinde gelişir. 8. haftada orta kulak yarığı oluşur ve kavite 30. haftada tamamlanır. İlk ark kıkırdağı, gebeliğin 4. ayında kemikleşmeye başlayan 8. gebelik haftasında malleus ve inkus oluşturur. İkinci kemerden kıkırdak, otik kapsülden türetilen ayak plakasının medial laminası dışında stapesten çıkar. 9. haftada, ektodermal hücreler çoğalır, meatus lümenini doldurur ve “meatal tıkaç” (MP) oluşturur. Daha sonra 10. haftada, MP yatay bir düzlemi takip ederek disk benzeri bir şekilde uzanır ve MP’nin iç kısmı gelecekteki timpanik membranı oluşturmak için incelmeye başlar.
Aynı zamanda boynun proksimal kısmındaki tıkaç yeniden emilmeye başlar. 13. haftada MP, primordial malleus ile temas halindedir ve bu temas, 15. haftada timpanik zarı oluşturacak olan MP’nin iç kısmının inceliğine katkıda bulunacaktır. 16. haftada dış kulak kanalı tamamen açık ancak yine de dardır ve kavisli. 18. haftada meatus tamamen uzatılır ve 28. haftada tamamlanacak olan açılışına başlar.
4. ve 25. gebelik haftalarında meydana gelen ve bu gelişmelerden birini kesintiye uğratan her türlü advers olay, farklı CAA türlerinden birinden sorumlu olabilir. Advers olay, genetik anormallikler, vasküler kaza (fetal hipoksi), teratojenik maddeler (aminoglikozid antibiyotikler, hidantoin, alkol, nikotin, herbisitler), maternal enfeksiyon (kızamıkçık, Sitomegalovirüs, kızamık, hepatit, toksoplazmoz, lues) ve anneye bağlı olabilir. metabolik hastalık (tiroid hormonu eksikliği veya diyabet). CAA tek bir malformasyon olabilir veya oto-fasiyal disostoz (Treacher-Collins sendromu, Goldenhar sendromu), kraniofasiyal disostoz (Crouzon sendromu, Apert sendromu), oto-servikal disostoz (Klippel-Feil sendromu) durumunda olduğu gibi diğer malformasyonlarla ilişkili olabilir. , Wildervanck sendromu), oto-iskelet dizostoz (Van der Hoeve-De Klein sendromu, Albers-Schonberg hastalığı) ve kromozomal sendromlar (trizomi 13, 18, 21 ve 18q sendromu).
CAA Hastalarında BAHI Kullanma Endikasyonları
 CAA ağırlıklı olarak tek taraflıdır (yaklaşık % 70–90) ve malformasyon çoğunlukla sağ kulağı etkiler, belki de bu taraf kalbin bulunduğu ve genellikle bir baskıya sahip olan sol taraf yerine daha sık hipoperfüzyondan muzdarip olabilir 10 Sağ tarafa doğru mmHg daha yüksek. Kulak malformasyonlarının görülme sıklığı yaklaşık 3800 yenidoğanda 1’dir. Bazı çocuklar malformasyon izole bir hastalık olmadığında bilateral bir CAA sergileyebilir, ancak bir sendromda örneğin CHARGE sendromu gibi bağlamsallaştırılır, burada çocukların bilateral atreziden % 60’a kadar etkilendiği bir durumdur.
CAA ağırlıklı olarak tek taraflıdır (yaklaşık % 70–90) ve malformasyon çoğunlukla sağ kulağı etkiler, belki de bu taraf kalbin bulunduğu ve genellikle bir baskıya sahip olan sol taraf yerine daha sık hipoperfüzyondan muzdarip olabilir 10 Sağ tarafa doğru mmHg daha yüksek. Kulak malformasyonlarının görülme sıklığı yaklaşık 3800 yenidoğanda 1’dir. Bazı çocuklar malformasyon izole bir hastalık olmadığında bilateral bir CAA sergileyebilir, ancak bir sendromda örneğin CHARGE sendromu gibi bağlamsallaştırılır, burada çocukların bilateral atreziden % 60’a kadar etkilendiği bir durumdur.
Hastalar farklı şiddette dış ve orta kulak malformasyonundan etkilenebil. Ciddiyet nedeniyle farklı işitme kaybı formlarını belirlenebilir. CAA tipik olarak vakaların % 80-90’ında iletim tipi işitme kaybına (CHL) neden olurken, geri kalan hastalarda sensörinöral işitme kaybı (SNHL) bileşenidir. CHL tipik olarak 40-60 dB’lik orta düzeyde işitme kaybı aralığındadır; bu, BAHI’lerin daha iyi çalıştığı aralıktır.
Arkadaki görüntü, BAHI’yi kullanmak için ideal koşulu gösterirken, süper etkilenen sarı muz, ses frekansının dağılımını gösterir. 45 dB içinde bir işitsel eşik sunan CHL, bir BAHI’den faydalanabilir çünkü implant, 500 ile 4000 Hz aralığında işitsel işlevlerin iyi bir şekilde kurtarılmasını garanti eder. Tek taraflı CAA’dan ve sensörinöral işitme kaybından (CAA’lı çocukların % 10-20’si) muzdarip çocuklar söz konusu olduğunda, kontralateral normal işitme fonksiyonu korunursa, BAHI işitme fonksiyonunu eski haline getirmek için kullanılabilir.
Kaynakça:
https://pubmed.ncbi.nlm.nih.gov/23851888/
https://www.stanfordchildrens.org/en/service/hearing-center/conditions/aural-atresia-microtia
https://texasearcenter.com/disorders-of-hearing/congenital-aural-atresia
Yazar: Özlem Güvenç Ağaoğlu