
Huntington Koresi olarak da bilinen Huntington hastalığı, beyin hücrelerinin ölümüyle sonuçlanan, nörodejeneratif, kalıtsal bir hastalıktır. İlk semptomlar genellikle duygudurum ve zihinsel yeteneklerde görülen problemlerdir. Genel bir koordinasyon eksikliği, kararsız bir yürüyüş söz konusudur ve hastalık ilerledikçe sarsıntılı vücut hareketleri daha belirginleşir. Fiziksel beceriler, eşgüdümlü hareket imkansızlaşacak ve kişi konuşamayacak hale gelecek kadar zayıflar. Zihinsel yeteneklerdeki çöküş demansa dönüşür. Her yaşta başlayabilirse de, semptomlar çoğunlukla 30 ila 50 yaş arasında başlar. Hastalık, her ardışık kuşakta hayatın daha erken dönemlerinde gelişebilir özelliktedir. Vakaların yaklaşık yüzde 8’i 20 yaşından önce başlar ve genellikle Parkinson hastalığına benzer semptomlarla kendini gösterir. Huntington vakaları genellikle sorunlarının derecesini hafife alırlar.
Huntington Hastalığının Nedeni Nedir?![Nörodejeneratif Huntington Hastalığının Tedavisi Var mı?]()

Huntington, tipik olarak kişinin ebeveyninden miras kalmaktadır. Ancak, olguların yüzde 10’unda yeni bir mutasyon görülmektedir. Hastalığa, vakanın “Huntingtin” adlı geninin iki kopyasındaki otozomal dominant bir mutasyon neden olmaktadır. Bu, kişinin çocuğunun genellikle hastalığın yüzde 50 oranında bir kalıtım olasılığına sahip olduğu anlamına gelmektedir. Huntingtin geni, “avlayan” olarak da adlandırılan bir protein için genetik bilgi sağlamaktadır. Huntingtin proteinini kodlayan gen içindeki CAG (sitozin-adenin-guanin) üçlü tekrarlarının yayılması, anormal bir protein ortaya çıkarır ve bu protein tam olarak anlaşılamamış mekanizmalarla yavaş yavaş beyindeki hücrelere zarar verir. Teşhis, genetik test ile yapılmaktadır ve semptomların var olup olmamasına bakılmaksızın, herhangi bir zamanda gerçekleştirilebilir.
Huntingtin proteini, 100’ün üzerinde proteinle etkileşime girmektedir ve çoklu biyolojik fonksiyonlara sahip görünmektedir. Bu mutasyon geçiren proteinin davranışı tam olarak anlaşılamamıştır, ancak özellikle beyindeki bazı hücre tiplerine toksik etkisi olduğu bilinmektedir. İlk verdiği hasar, en çok striatumda görülür. Ancak hastalık ilerledikçe beynin diğer alanları da belirgin bir şekilde etkilenir. Erken semptomlar, striatumun fonksiyonları ve kortikal bağlantıları, yani hareket kontrolü, duygudurum ve yüksek bilişsel işlevlerdir. Bilinen nöropsikolojik belirtiler, anksiyete, depresyon, duygu sığlığı, saldırganlık, benmerkezcilik ve kompulsif davranışlardır. Bunlar alkolizm, kumar ve hiperseksüalite gibi bağımlılıklara neden olabilmektedir. Diğer insanların olumsuz ifadelerini tanımada güçlükler de gözlemlenmektedir.
DNA’nın metilasyonu da Huntington’da değişiyor gibi gözükmektedir.
Hastalığın henüz herhangi bir tedavisi yoktur. İleri evrelerinde tam zamanlı bakım gereklidir. Müdaheleler bazı belirtileri kısmen rahatlatabilmekte ve yaşam kalitesini iyileştirebilmektedir. Hareket problemlerinin tedavisinde en iyi sonuç tetrabenazin ile alınmaktadır. Huntington, Avrupa kökenli 100.000 kişide yaklaşık 4 ila 15 kişiyi etkilemekte, Japonlar arasında nadir olarak görülmekte, Afrika’daki oranı ise bilinmemektedir. Hastalık, erkekleri ve kadınları eşit derecede etkilemekte, pnömoni, kalp rahatsızlığı ve düşmelerden kaynaklanan fiziksel yaralanmalar gibi komplikasyonlar yaşam beklentisini azaltmaktadır. Vakaların yaklaşık yüzde 9’unda ölüm nedeni intihar olmaktadır. Ölüm, tipik olarak, hastalığın ilk tespit edildiği tarihten itibaren on beş ila yirmi yıl arasında gerçekleşmektedir.
Hastalığın ilk tanımlanması Charles Oscar Waters tarafından 1841’de yapıldı. Tanım, daha sonra 1872’de George Huntington tarafından daha ayrıntılı bir şekilde genişletildi. Genetik temeli, 1993 yılında, Kalıtsal Hastalıklar Vakfı liderliğindeki uluslararası bir ortak araştırma ile keşfedildi. 1960’ların sonlarında, halkı bilgilendirmek, hasta ve ailelerine destek sağlamak ve yeni araştırmalar yapmak için araştırma ve destek örgütleri kurulmaya başladı. Günümüzdeki araştırmalar, hastalığın mekanizmasını ayrıntılı olarak belirlemek, araştırmalara yardımcı olacak hayvan modellerinin mühendisliğini geliştirmek, semptomları tedavi edecek yeni ilaç deneyleri yapmak, hastalığın ilerlemesini yavaşlatmak ve hastalığın neden olduğu hasarı onarmak amacı ile kök hücre tedavisi geliştirmek gibi prosedürlere odaklanmaktadır.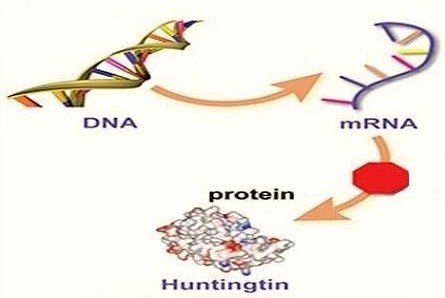
Huntington hastalığının yıkıcı etkilerinden sorumlu proteinin düzeyini düşürmek için tasarlanmış bir ilacın güvenlirliğini insanlar üzerinde inceleyen araştırmalardan başarılı sonuçlar alındı. Sevinmek için erken olsa da, heyecan verici bu sonuçlar, Alzheimer ve Parkinson gibi diğer beyin bozukluklarının da tedavilerine uyarlanabilecek ilacın potansiyelini ortaya koyuyor. HTTRx kodlu bir ilaç, mutasyona uğramış gen tarafından gönderilen mRNA sinyallerini engelliyor. Bozulan proteinin üretimini etkili bir şekilde durduruyor, Huntingon’un ilerlemesini yavaşlatıyor ya da başlamasını önlüyor.
HTT Geni ve Huntingtin Proteini Nedir?
HTT geni, huntingtin adlı proteinin üretilmesi talimatlarını sağlamaktadır. Huntingtin proteininin işlevleri tamamen bilinmemekle birlikte, beynin sinir hücrelerinde (nöronlarda) önemli bir rol oynamaktadır ve doğumdan önceki normal gelişim için gereklidir. Huntingtin, vücut dokularının çoğunda, ayrıca beyinde en yüksek aktivite seviyesine sahip olarak bulunur. Bu protein, hücrelerdeki kimyasal sinyalizasyon, materyallerin nakledilmesi, diğer proteinlere ve diğer yapılara bağlanma ve hücreyi kendini tahrip etmesinden (apoptozdan) koruma işlevleriyle ilgilidir. HTT geninin bir bölümü, bir CAG trinükleotid yinelenme olan belirli bir DNA parçasını kapsar. Bu segment, üst üste birden çok kez görünen üç DNA yapı bloğu (sitozin, adenin ve guanin) serisinden oluşur. CAG segmenti genin içinde normal olarak genellikle 10 ila 35 kez tekrarlanmaktadır.
Huntington hastalığına neden olan kalıtsal mutasyon, CAG trinükleotid segmentinin tekrar sayısının artması olarak bilinir. Mutasyon, HTT genindeki CAG segmentinin boyutunu arttırmaktadır ve Huntington hastalığı olanlarda CAG tekrarları 36’dan 120’ye kadar çoğalmaktadır. 36 ila 39 CAG tekrarı, insanların Huntington hastalığının belirti ve semptomlarını geliştirme riskini başlatmakta, 40 veya daha fazla tekrar hastalığın oluşmasını kesinleştirmektedir.
Genişleyen CAG segmenti, huntingtin proteininin anormal şekilde uzun bir versiyonunun üretilmesine yol açmaktadır. Uzayan protein küçük, toksik parçalara ayrılır. Toksik parçaların oluşturduğu kümeler nöronlarda birikerek, bu hücrelerin normal işlevlerini bozar. Bu süreç özellikle hareketin koordinasyonunda ve düşünce ve duyguların kontrolünde önemi olan beynin striatum ve serebral korteks bölgelerini etkiler. Huntington hastalığının belirtilerinin altında nöronların işlev bozukluğu ve ölümü yatar.
Mutasyona uğramış HTT geni bir nesilden diğerine geçtikçe, CAG trinükleotid tekrarının boyutları artar. Daha çok tekrar, genellikle belirtilerin erkenden ortaya çıkmasına yol açmaktadır. Huntington hastalığının yetişkinlik döneminin ortalarında görülen yetişkin başlangıçlı formunda tipik olarak HTT geninde 40 ila 50 CAG tekrarı görülürken, çocukluk çağında veya ilk gençlik döneminde görülen bozukluğun çocuksu formunda 60’dan fazla CAG tekrarı görülmektedir. HTT geninde 27 ila 35 CAG tekrarı olan kişiler Huntington hastalığına yakalanmamakla birlikte, bozukluğu geliştirecek çocuk sahibi olma riski altındadırlar. Gen ebeveynden çocuğa geçtikçe, CAG trinükleotid tekrarının boyutu Huntington hastalığına (36 veya daha fazla tekrar) eşlik eden aralığa kadar uzayabilir.
HTTRx Nedir?![Nörodejeneratif Huntington Hastalığının Tedavisi Var mı?]()

HTTRx koduyla bilinen ilaç, HTT geni ve huntingtin proteini arasında, proteinin üretimini önlemek için bir ara madde olarak etkileşime giren antisens bir ilaçtır. Spinal kordun alt bölümüne, lomber omurgaya intratekal enjeksiyon olarak bilinen yöntemle L3-L4 aralığından girilerek enjekte edilmektedir. İlaç, kan-beyin bariyeri olarak bilinen beyin ve kan arasındaki arayüzü geçemediği için oral olarak alınamamaktadır. Kan-beyin bariyeri birçok terapinin gelişimi için de bir problem teşkil etmektedir.
On yıllık bir geliştirme sürecini ve hayvanlar üzerindeki başarılı denemeleri takiben, insanlar üzerindeki çalışmaların ilk aşaması, ilacın tedavi için umutlandırıcı olduğunu gösteriyor. İnsanlar üzerinde yapılan herhangi bir araştırmanın ilk aşamasında, her zaman öncelikle ilacın etkinliği değil, insan sağlığına herhangi bir zararı olmadığını kanıtlamak önem taşımaktadır. Araştırmacılar, ilacın iyi tolere edildiğini bildirdiler. İlk denemelerin birincil hedefi olmasa da, ilacın etkinliği de, araştırmacılar tarafından olumlu ve heyecan verici olarak kaydedildi. İlaç tedavisinden sonra, deneklerin omurilik sıvısında ölçülen mutasyona uğramış protein varlığında önemli bir azalma olduğu kanıtlandı.
Deneyler, İngiltere, Almanya ve Kanada’daki dokuz çalışma merkezinde Huntington hastası 46 vakayla sürdürüldü. Her hastanın beynine erişmesini sağlamak üzere, omurilik sıvılarına enjekte edilen dört doz HTTRx veya plasebo verildi. Faz 1 / 2a denemeleri ilerledikçe, HTTRx’in dozu, artan doz deneyi tasarımına göre birkaç defa arttırıldı. Hastaların güvenliği çalışma boyunca bağımsız bir güvenlik komitesi tarafından izlendi. Ultra-duyarlı tahlil cihazları kullanılarak, tedavi öncesinde ve sonrasında her hastanın omurilik sıvısındaki protein konsantrasyonları dikkatle ölçüldü. HTTRx, mutant huntingtin seviyesinin doza bağlı olarak önemli ölçüde düşürülmesini sağladı.
Sonuç
Londra Üniversitesi Akademisi (UCL) Nöroloji Enstitüsü araştırmacılarından ve yönettiği bu çalışmayla 2017 Uluslararası Leslie Gehry Brenner Ödülü’nü alan Klinik Nöroloji Profesörü Sarah Tabrizi, insanlar üzerinde 2015 yılında denenmeye başlanan ilaçla ilgili araştırma sonuçlarının Huntington hastaları ve aileleri için büyük bir öneme sahip olduğunu ve ilk kez bir ilacın sinir sistemindeki toksik hastalıklara neden olan protein düzeyini düşürdüğünü vurguluyor. Ionis-HTTRx deneyleri, randomize, çift-kör, plasebo kontrollü uygulamalardı. Özetle, hastaların ve doktorların hangi hastaya gerçek ilacın verildiğini bilmedikleri, böylece, tedavinin kendisinin değil, hastanın tedaviye olan inancından dolayı iyileşme hissi riskini yani plasebo etkisini ortadan kaldıran çalışmalardı. RG6042 olarak yeniden adlandırılan ilaçla sürdürülen araştırmaların belki de en önemli sonucu, ilacın diğer nörodejeneratif hastalıklar için de öngörülen potansiyel uygulamalar olacaktır. İşlevsel olmayan proteinler, Alzheimer ve Parkinson gibi hastalıklarla bağlantılıdır ve bu ilaç, söz konusu proteinlerin etkin bir şekilde hedeflenebileceği ve üretimlerinin azaltılabileceği bir sürecin başlangıcıdır.
Kaynakça:
-Sarah Tabrizi, et al, “Drug lowers deadly Huntingtons disease protein”, University College London, UCL news, (2017).
-P.Dayalu, R.L.Albin, “Huntington disease: pathogenesis and treatment”, Neurologic Clinics, 33 (1), (2015).
-E.van Duijn, E.M.Kingma, R.C.van der Mast,”Psychopathology in verified Huntington’s disease gene carriers”, J Neuropsychiatry Clin Neurosci. 19 (4), (2007).
Yazar: Oben Güney Saraçoğlu