
Akonddrogenezis, gövde, kol, bacak, anormal kaburga, omur ve diğer iskelet anormalliklerine göre aşırı kısalması ile karakterize nadir bir iskelet displazisi grubudur. Bu durumlarla ilişkili sağlık sorunları yaşamı tehdit eder ve etkilenen bebeklerin çoğu doğumdan kısa bir süre sonra doğarlar veya solunum yetmezliği nedeniyle ölürler. Tüm akondrogenezis türleri genetik koşullardır; tip IA ve tip IB otozomal resesif bozukluklardır, akonddrogenezis ise tip II otozomal dominant bir hastalıktır. Tüm akondrogenezis tipleri, genellikle doğum öncesi 14-17. Haftalar gibi erken doğum öncesi ultrason muayenesi ile saptanan iskelet displazileridir.
Akonddrogenezis terimi ilk olarak 1952 yılında tıp literatüründe Marco Fraccaro adlı bir İtalyan patolog tarafından kullanılmıştır. Kelime Yunanca’dan türemiştir ve “kıkırdak üretmemek” anlamına gelir. Anormal büyüme veya kıkırdak ve kemik gelişimi ile karakterize edilen bir grup hastalık için (yaklaşık 450 klinik tanı) geniş bir terim olan iskelet displazileri grubuna (osteokondrodisplaziler olarak da adlandırılır) aittir.
Belirtileri
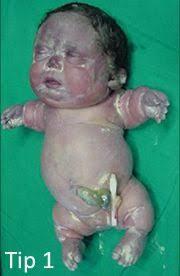
Akonddrogenezis, erken doğum, vücutta anormal sıvı birikimi (hidrops fetalis) ve şekli anormal olabilen ve daha az ossifiye olmuş bir kafa ile karakterize edilir. Gövde küçük olduğu için kafa orantısız şekilde büyük görünebilir. İlaveten, etkilenen bireylerde son derece kısa uzuvlar ve kaburgalar, kısa boyunlu, düz omurlar ve iskeletin diğer birçok kemikleri uygun şekilde gelişmemiştir. Bu bozuklukla doğan bebeklerde karın belirgindir ve torasik kafes küçüktür. Diğer anormallikler ağız çatısının (yarık damak), korneanın bulanıklaşması ve kulak deformitelerinin tam kapanmamasıdır. Bozukluk, doğumdan önce veya doğumdan kısa bir süre sonra genellikle az gelişmiş toraks ve küçük akciğerler nedeniyle hayatı tehdit eder. Belirtileri tiplerine göre farklılıklar gösterir. 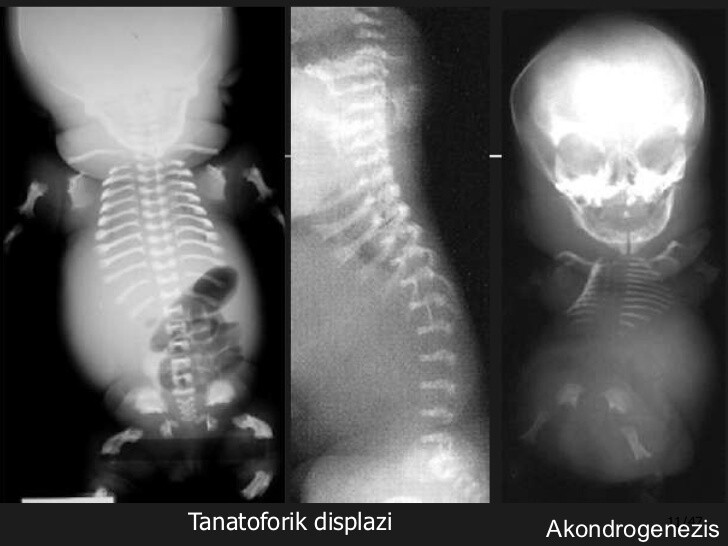
Akonddrogenezis tip IA (Houston-Harris tipi): Değişken yüz anormallikleri (düz yüz, çıkıntılı gözler ve çıkıntılı dil veya sadece küçük yüz anomalileri), kısa gövde ve uzuvlar, kısa kaburgalar ve ince kafatası kemikleri (kafatasının eksik kemik yapısı) ile karakterize edilir. Omurga, pelvis ve ekstremitelerde kemik oluşumu anormaldir, ancak iskelet tutulumunun ciddiyet derecesi değişebilir. Ancak, küçük göğüs kafesi akciğerlerin az gelişmesine ve doğumdan kısa bir süre sonra ölüme yol açmaktadır.
Akonddrogenezis tipi IB (Fraccaro tipi): Hastalığın bu tipi kısa gövde ve uzuvlar, dar göğüs ve belirgin karın ile karakterizedir. Etkilenen bebeklerde göbek deliğinin etrafında (göbek fıtığı) veya kasıkların yakınında (kasık fıtığı) bir çıkıntı olabilir ve ayakları içe dönük, kısa ayak parmaklarına sahip olabilir. Yüz düz olabilir, damak yarık olabilir ve boyun genellikle kısadır. Bazı durumlarda, boynun yumuşak dokusu anormal şekilde kalınlaşabilir Akonddrogenezis tip IB, SLC26A2 genindeki mutasyonlarla ilişkili küçük bir grup bozukluk olan bir sülfasyon bozukluğu olarak alt sınıfa ayrılır. Bu grup diastrofik displaziyi ve ılımlı koşullar olan resesif multipl epifiz displazisini içerir. Genetik değişiklikler aynı gende olsa bile, bebek gelişiminde bir tanının diğerine değişmediğine dikkat etmek önemlidir.
Akonddrogenezis tip II (Langer-Saldino tipi): Dar bir göğüs, kollarda ve / veya bacaklarda anormal derecede küçük veya kısa kemikler, ince kaburgalar, düz omurlar veya vertebral cisimlerin, az gelişmiş akciğerler, küçük çene, yarık damak ve yetersiz kemik salınımı ile karakterizedir. Ayaklar, omurga ve pelviste kemik oluşumu anormaldir. Anormal sıvı birikimi meydana gelebilir (hidrops fetalis) ve karın genişleyebilir.
Nedenleri
Her tip akonddrogenezis, spesifik bir gendeki bir mutasyondan kaynaklanır. Genler, vücudun birçok fonksiyonunda kritik bir rol oynayan proteinlerin oluşturulması için talimatlar sağlar. Bir genide mutasyon meydana geldiğinde, protein ürünü hatalı, yetersiz veya eksik olabilir. Belirli bir proteinin fonksiyonlarına bağlı olarak, bu vücudun birçok organ sistemini etkileyebilir.
Akonddrogenezis tip IA ve IB tipine neden olan gen mutasyonları otozomal resesif şekilde kalıtsaldır. Resesif genetik bozukluklar, bir birey aynı özellik için anormal bir genin iki kopyasını her bir ebeveynden bir tane aldığında ortaya çıkar. Bir birey hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için taşıyıcıdır, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin etkilenen birinin hamileliğinde çocuğun bu hastalığa sahip olma riski her hamilelikte % 25’tir. Çocuğun ebeveynleri gibi taşıyıcı olma riski her hamilelikte % 50’dir. Bir çocuğun her iki ebeveynden de normal gen alma şansı% 25’tir. Risk erkeklerde ve kadınlarda aynıdır. Tüm bireyler birkaç anormal gen taşır. Yakın akraba olan ebeveynler (akraba), akraba olmayan ebeveynlerden daha nadir bir resesif genetik bozukluğu olan çocuklara sahip olma riskini artıran aynı anormal geni taşıma şansına sahiptir.
 Akonddrogenezis tip IA, TRIP11 genindeki mutasyonlardan kaynaklanır. Akonddrogenezis tip IB, SLC26A2genindeki mutasyonlardan kaynaklanır. Bu iki gen, iskelet ve diğer dokular oluşturmak için gereken belirli kıkırdak proteinlerinin verimli hücresel nakli için gereklidir. TRIP11 geninin mutasyonları, Golgi mikrotubule bağlı protein 210’un eksikliğine yol açar. Bu protein, vücudun çoğu hücre tipinde bulunur. SLC26A2 geninin protein ürünü, kıkırdakta doğru gelişme ve işlevde rol oynayan bir sülfat taşıyıcıdır. Kıkırdak, eklemlerde tampon görevi gören özel dokudur. Bir embriyonun iskeletinin çoğu yavaş yavaş kemiğe dönüşen kıkırdaktan oluşur.
Akonddrogenezis tip IA, TRIP11 genindeki mutasyonlardan kaynaklanır. Akonddrogenezis tip IB, SLC26A2genindeki mutasyonlardan kaynaklanır. Bu iki gen, iskelet ve diğer dokular oluşturmak için gereken belirli kıkırdak proteinlerinin verimli hücresel nakli için gereklidir. TRIP11 geninin mutasyonları, Golgi mikrotubule bağlı protein 210’un eksikliğine yol açar. Bu protein, vücudun çoğu hücre tipinde bulunur. SLC26A2 geninin protein ürünü, kıkırdakta doğru gelişme ve işlevde rol oynayan bir sülfat taşıyıcıdır. Kıkırdak, eklemlerde tampon görevi gören özel dokudur. Bir embriyonun iskeletinin çoğu yavaş yavaş kemiğe dönüşen kıkırdaktan oluşur.
Akonddrogenezis tip 2’ye neden olan gen mutasyonu, COL2A1 geninde otozomal dominant değişim olarak adlandırılır. Baskın genetik bozukluklar, belirli bir hastalığa neden olmak için anormal bir genin sadece bir kopyasının gerekli olduğu durumlarda ortaya çıkar. Çoğu tip akonddrogenez vakası, COL2A1’deki yeni ( de novo ) mutasyondan kaynaklanmaktadır. Bu, etkilenen bir bebeğin ebeveynleri için aynı hastalığı olan başka bir çocuğu alma riskinin% 1’den daha yüksek olmadığı anlamına gelir. Bu gen, kollajen tip II’yi kodlamaktadır. Kolajen vücutta en bol bulunan proteinlerden biridir ve doku formunu ve gücünü veren, vücuttaki hücreler arasındaki materyal olan bağ dokusunun büyük bir yapı bloğudur. Romen rakamları ile gösterilen birçok farklı kollajen tipi vardır. Kolajen tip II, en sık kıkırdakta ve gözün merkezini dolduran jöle benzeri sıvıda (vitröz) görülür. Kolajen ayrıca kemikte bulunur.
Akonddrogenez tip II olan bebeklerin kardeşlerinin etkilendiği çok nadir vakalar vardır. Bu, büyük olasılıkla yumurtada veya spermde bir ebeveynden (germline mozaikliği) birden fazla hücre çizgisinin varlığından kaynaklanmaktadır. Germline mozaiklemede, bir ebeveynin üreme hücrelerinin bazıları (germ hücreleri) COL2A1 gen mutasyonunu taşır, diğer germ hücreleri normal COL2A1 genlerini içerir. Ebeveynin vücudundaki diğer hücrelerde mutasyon yoktur, bu nedenle bu ebeveynler etkilenmez. Sonuç olarak, ebeveynlerin çocuklarından biri veya birkaçı, COL2A1 germ hücre genini miras alabilirler. Ana akordrojenez II gelişmesine yol açan mutasyon, ebeveynde bu bozukluk yoktur Germline mozaikçiliğinin, görünüşte etkilenmeyen ebeveynlerin aynı otozomal dominant genetik hastalığı olan birden fazla çocuğa sahip olması durumunda şüphelenilebilir. Çocuğa mozaik germ hattı mutasyonundan geçen bir ebeveynin olasılığı, ebeveynin mutasyona sahip olan germ hücrelerinin yüzdesine göre olmayan yüzdesine bağlıdır. Gebelikten önce germline mutasyon testi yoktur. Hamileliğin erken döneminde yapılan testler mümkün olabilir ve en iyi şekilde doğrudan bir genetik uzmanıyla tartışılabilir.
Etkilenen Kişiler
Akonddrogenezis erkekleri ve dişileri eşit sayıda etkiler. Akonddrogenezis tip IA ve tip IB çok nadir görülen bozukluklardır ve prevalansı bilinmemektedir. Achondrogenesis tip II, yaklaşık 1/40,000-1/60,000 yeni doğanlarda meydana gelir.
İlgili Bozukluklar
İskelet displazileri (osteochondrodysplasias): Anormal büyüme, gelişme veya kıkırdak ve kemik ile karakterize edilen bir grup hastalık için genel bir terimdir. Bazı formlar doğumdan kısa bir süre sonra hayatı tehdit eden komplikasyonlara neden olurken, diğerleri sadece yaşamı tehdit eden komplikasyonlara neden olabilir veya olmayabilir. Bazı formlar, yaşamın erken döneminde hayatı tehdit eden komplikasyonlara neden olmaz. İskelet displazileri, kısa ekstremeli kısa boylarla veya gövde ve uzuvların daha orantılı olarak kısalmasıyla ilişkilendirilebilir. Spesifik bozukluğa bağlı olarak çeşitli ek anormallikler bulunabilir. 360’dan fazla nedensel gen içeren yaklaşık 450 çeşit iskelet displazisi vardır. Birkaç form aşağıda tartışılmıştır; aralarındaki fark radyografik ve moleküler yollarla yapılır.
Sınırlı displaziler: COL2A1 genindeki bir değişimin (mutasyonun) neden olduğu birkaç iskelet displazisi formundan biridir. Bu gen, kemiklerin ve diğer bağ dokularının normal gelişimi için gerekli olan kollajen tip II’yi oluşturan belirli bir proteinin üretiminde rol oynar. Kollajen tip II’nin kompozisyonundaki değişiklikler anormal iskelet büyümesine ve dolayısıyla iskelet displazileri olarak bilinen çeşitli konjenital iskelet hastalıklarına yol açar. Kısa boy, geniş dizler ve yarık damak gibi Kniest displazinin belirti ve semptomlarından bazıları genellikle doğumda bulunur. Diğer semptomlar yaşla birlikte gelişir.
Kampomelik sendromu: Çoklu anomalilerin bulunduğu nadir bir doğuştan hastalıktır. SOX9 genindeki mutasyonlardan kaynaklanır ve bacakların, özellikle tibiaların uzun kemiklerinin eğilmesi ve açısal şekli ile karakterize edilir. Yüzün çoklu minör anomalileri; yarık dudaki, omuz ve pelvik bölgenin anormallikleri ve normal on iki yerine onbir kaburga çifti gibi diğer iskelet anomalileri bulunur. Ayrıca trakeanın az gelişmişliği bazı vakalarda gelişimsel gecikme ve kadınlarda göründüğü gibi erkeklerde yetersiz cinsel organ gelişimi görülür.
Hipofosfatazi: Raşitizme benzeyen iskelet defekti ile karakterize kemiklerin doğuştan metabolik bir hastalığıdır. Belirtiler, kemik mineralinin genç, kireçlenmemiş kemikte (uzun yıllar sonunda uzun kemiklerin (epifizler) sonunda kıkırdakta birikmemesi sonucu ortaya çıkar. Kan serumunda ve kemik hücrelerinde alkalin fosfataz enziminin etkinliği normalden düşüktür. Fosfoetanolamin ve inorganik pirofosfatın idrarla atılma ve kan plazma konsantrasyonları anormal derecede yüksektir. Hipofosfatazi, Strensig ile tedaviye yanıt  verir.
verir.
Tanatoforik displazi: Ölümcül iskelet displazilerinin en sık görülen formlarından biridir. Büyümüş kafa, kollarda ve bacakların kısa olması, yaylı kemikler, küçük, kısa kaburgalar, dar toraks ve düzleştirilmiş omurlar ile karakterizedir. Anormal derecede büyük miktarda amniyotik sıvı var. Tanatoforik displazi FGFR3 genindeki mutasyonlardan kaynaklanır.
Kısa Kaburga Polidaktili Sendromu: Birkaç tip iskelet displazisi bulunur. Bebeğin yarık dudak ve damak, deforme olmuş kulaklar, kısa kaburgaları olan dar bir göğsü olabilir. Böbrekler, cinsel organları sıklıkla deforme olur. Beyin malformasyonu ve bir safra kesesi yokluğu olabilir. Bu hastalık yetersiz akciğer gelişimi sonucu yaşamı tehdit edicidir.
Teşhis
Akonddrogenezis, fiziksel özellikler, röntgen (radyografik) bulgular ve doku örneklerinin mikroskopta incelenmesi (histoloji) ile teşhis edilir. SLC26A2 genindeki mutasyonlar için moleküler genetik testler, 1B akonddrogenez teşhisini doğrulamak için kullanılabilir. Akonddrogenezin ultrason ile prenatal tanısı 14-15. Haftalar arasında mümkündür. Bir aile üyesinde spesifik gen mutasyonları tanımlanmışsa, koryonik villus örneklemesi (10-12 hafta gebelik) veya amniyosentez (15-18 hafta gebelik) ile prenatal tanı mümkündür.
Tedavi
Akonddrogenezis tedavisi semptomatiktir ve destekleyicidir. Bu tedavi doktorların ağrı, stres ve hastalıkla ilişkili spesifik semptomları azaltmaya veya en aza indirmeye çalıştıkları palyatif bakımı içerir. Etkilenen çocuğu olan ailelere genetik danışma önerilmektedir. Tüm aile için psikososyal destek de şarttır.
Kaynakça:
niams.nih.gov
mayoclinic.org
lupusresearch.org
Yazar: Özlem Güvenç Ağaoğlu