
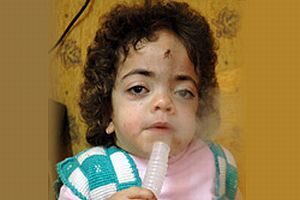
 MPS I (Hurler sendromu veya mukopolisakaridosis tip 1), kromozom 4’te mutasyona uğramış genlerin neden olduğu lizozomal enzim eksikliğine neden olan metabolik bir bozukluktur. Sendroma genellikle genç bebeklerde (3-6 aylıkken) teşhis konur.MPS I’in birçok belirti ve semptomu vardır.İlk belirtileri; genellikle ağız genişlemiş, kalın dudakları ve giderek kötüleşen göz problemleriyle birlikte yüz özelliklerini sertleştirmektedir .
MPS I (Hurler sendromu veya mukopolisakaridosis tip 1), kromozom 4’te mutasyona uğramış genlerin neden olduğu lizozomal enzim eksikliğine neden olan metabolik bir bozukluktur. Sendroma genellikle genç bebeklerde (3-6 aylıkken) teşhis konur.MPS I’in birçok belirti ve semptomu vardır.İlk belirtileri; genellikle ağız genişlemiş, kalın dudakları ve giderek kötüleşen göz problemleriyle birlikte yüz özelliklerini sertleştirmektedir .
Olabilecek diğer belirtileri şunlardır:
Büyütülmüş kafa
Kronik burun akıntısı
Retinal dejenerasyon
Büyümüş karın organları
Splenomegali
Iskelet displazisi
Eklem sertliği
MPS I’in nedeni lizozomal enzim aL-iduronidazın eksikliğine yol açan kromozom 4 üzerinde kalıtsal genetik mutasyonlardır.MPS I, her ebeveynden aldıkları otozomal resesif bir gen tarafından devralınır. MPS I, kan testleri ve spinal X-ışınları, ekokardiyografi ve semptomların ortaya çıktığı diğer testler gibi diğer testlerle birlikte bireyin erken klinik özellikleri ( örneğin, yüz biçiminin sertleşmesi ) ile teşhis edilir.
 MPS I için herhangi bir tedavi mevcut değildir ve yönetilmesi (tedavisi) zordur. Laronidaz (Aldurazyme) ile tıbbi tedavi ve muhtemelen bu kalıtsal hastalığın semptomlarının bazılarını azaltmaya yardımcı olan cerrahi yöntemler kullanılır.
MPS I için herhangi bir tedavi mevcut değildir ve yönetilmesi (tedavisi) zordur. Laronidaz (Aldurazyme) ile tıbbi tedavi ve muhtemelen bu kalıtsal hastalığın semptomlarının bazılarını azaltmaya yardımcı olan cerrahi yöntemler kullanılır.
MSP I’li bir kişinin yaşam beklentisi mutasyonların ciddiyetine göre değişir. En yumuşak formu olan insanlar (MPS İS) oldukça normal bir ömre sahip olabilir, ılımlı formu olanlar (MPS IH / S) ergenlik çağındayken veya erken erişkinlikte yaşayabilir, ancak en şiddetli form olan MPS IH veya Hurler sendromu ömrü nadiren 10 yıldan uzun olduğu görülür.
MPS I ( mukopolisakaridozis tip 1 ) Nedir?
MSP I (mukopolisakaridosis tip 1) lizozomal enzimdeki eksikliklerin neden olduğu metabolik bozukluğun en yaygın türüdür. Lizozom enzimleri, birçok farklı türde maddeyi parçalamaya yarayan organellerin, lizozom hücrelerinde bulunur.
MSP I, kromozom 4 üzerinde alfa-L-iduronidaz kodlayan genteki mutasyonlardan kaynaklanır ve bu da bağ dokusunun ana bir unsuru olan glikosaminoglikanın bozunmasına ihtiyaç duyan eksik lizozomal enzimlere neden olur.
Bu gende birçok farklı mutasyon vardır. Çeşitli mutasyonlar MPS IH (Hurler sendromu), MPS IS (Scheie sendromu) ve MPS IH / S (Hurler / Scheie sendromu) ve diğerlerine neden olur.
MPS I’in Belirtileri Nelerdir?
Daha önce belirtildiği gibi, MPS’nin belirtileri ve semptomları şiddet spektrumunu gösterebilir. MPS’nin belirtileri ve semptomlarından bazıları şunlardır:
 Yüz özelliklerinin kabalaştırılması (genellikle ilk 3-6 aylık anormallikler)
Yüz özelliklerinin kabalaştırılması (genellikle ilk 3-6 aylık anormallikler)
Kalın dudakları olan büyümüş ağız
Büyütülmüş kafa
Kronik burun akıntısı
Progresif kornea bulutlaması
Retinal dejenerasyon
Splenomegali
Inguinal ve umbilical herni
Karında büyümüş organlar
İskelet displazisi kısa boy ile sonuçlanabilir
Eklem sertliği
Kardiyak kapak hastalığı, özellikle aort kapağında
Sık Solunum Yolları Enfeksiyonları
Fonksiyonel yaş seviyesinin maksimum 2-4 yaşına kadar uzadığı gelişimsel gecikmeler
En şiddetli belirtiler MPS IH’de (Hurler sendromu) ortaya çıkar.MPS I Neden Ortaya Çıkar?
MPS I’nin nedeni, kromozom 4’te, lizozomal enzim aL-iduronidaz’ın eksikliğine ve dolayısıyla mukopolisakaritlerin doku ve organlarda birikmesine yol açan, bunların aktivitesine ve gelişimine müdahale eden genetik mutasyonlardır.
MPS Genetik Bir Hastalık Mıdır?
MPS I, otozomal resesif bir şekilde kalıtım gösterir. Sonuç olarak, MPS’li neredeyse tüm bireyler her ebeveynden resesif bir gen alıyor. Ebeveynlerin her ikisi de çekinik gene sahip olursa, ihtimal dörtte bir olup, bir çocuğun her ebeveynden bir otozomal resesif gen alacağı ve MPS I geliştirdiği söylenebilir.
MPS I İçin Hangi Uzmanlara Başvurulabilir?
Genellikle çocuk doktorları MPS I’ i ilk fark eden kişilerdir.Diğer alanlardan ise; genetikçiler, nörologlar, kardiyologlar, cerrahlar başvurulabilecek uzmanlardır.
MPS Nasıl Teşhis Edilir?
Klinik olarak, çocukluk çağı, 3-6 aylıkken, yüz özelliklerinde sertleşmeler görülürse, MPS I tanısından şüphelenebilir.
Laboratuvar testleri aşağıdakileri içerebilir:
Kan yaymalarındaki lenfositlerin, anormal sitoplazmik inklüzyonlar aramak üzere incelenmesi
Üriner glikozaminoglikan (gag) ölçümü
Periferik kan beyaz hücrelerinde al-iduronidaz ölçümü
Mevcut mutasyonun saptanması için DNA analizi
Diğer testler; omurga röntgeni, ekokardiyografi, işitme, görme ve gelişimsel düzen testlerini içerebilir.
MPS’nin Tedavi ve Yönetimi Nasıl ?
Laronidaz ile enzim replasman tedavisi, MSP I’li insanlarda yürüme ve solunum fonksiyonlarını artırabilir .
Uzmanlaşmış cerrahi bakım, hidrosefali azaltabilir ve korneal transplantasyon bazı hastalara yardımcı olabilir.
Bazı vakalarda solunumun iyileştirilmesi için kardiyovasküler hastalıklar için supap replasmanı yapılmış ve trakeostomiyapılmıştır.
Omurilik füzyonu , servikal omurgada eğriliğin daha da ilerlemesini önlemeye yardımcı olabilir ve karpal tünel serbestlemesi bazı hastalara yardımcı olmuştur.
Kaynakça:
https://www.medicinenet.com
Yazar: Merve Karaca