
Leigh sendromu nadir görülen bir genetik nörometabolik hastalıktır. Merkezi sinir sisteminin (yani, beyin, omurilik ve optik sinir) dejenerasyonu ile karakterizedir. Leigh sendromunun belirtileri genellikle üç ay ile iki yıl arasında başlar, ancak bazı hastalar birkaç yıl sonrasına kadar belirti ve semptom göstermezler. Belirtiler ilerleyici nörolojik bozulma ile ilişkilidir ve önceden edinilmiş motor becerilerinin kaybı, iştah kaybı, kusma, sinirlilik ve / veya nöbet aktivitesini içerebilir.
Leigh sendromu ilerledikçe, semptomlar ayrıca genel zayıflık, kas tonusu eksikliği (hipotoni) ve solunum ve böbrek fonksiyonlarının bozulmasına neden olabilecek laktik asidoz bölümlerini içerebilir. Genetik olarak belirlenmiş birkaç farklı enzim bozukluğu sendroma neden olabilir. Leigh sendromlu bireylerin çoğu, mitokondriyal solunum zinciri kompleksi veya piruvat dehidrojenaz kompleksi enziminin eksikliği gibi mitokondriyal enerji üretimi kusurlarına sahiptir. Çoğu durumda, Leigh sendromu otozomal resesif bir özellik olarak kalıtsaldır. Bununla birlikte, mitokondriyal bir DNA mutasyonundan dolayı X’e bağlı resesif ve maternal kalıtım, ek aktarım modlarıdır.
Belirtileri
Hızla ilerleyen bir nörolojik hastalık olan klasik Leigh sendromunun (infantil nekrotizan ensefalopati) belirtileri genellikle 3 ay ile 2 yaş arasında başlamaktaadır. Çoğu çocukta, ilk göze çarpan belirti, önceden edinilmiş motor becerilerinin kaybıdır. Erken başlangıçta (yani, 3 ay), baş kontrolünün kaybı ve kötü emme kabiliyeti ilk göze çarpan semptomlar olabilir. Buna derin bir iştah kaybı, tekrarlayan kusma, sinirlilik, sürekli ağlama ve olası nöbet aktivitesi eşlik edebilir. Gelişimsel kilometre taşlarına ulaşmada gecikmeler de meydana gelebilir. Etkilenen bebekler beklenen oranda büyümekte ve kilo alamamaktadır.
Leigh sendromunun başlangıcı çocuklukta daha sonra ise örneğin, 24 ayda, sözcükleri (dizartri) ekleyememe, yürüme veya koşma (ataksi) gibi istekli hareketleri koordine etmede zorluk yaşayabilir. Önceden edinilmiş zihinsel beceriler azalabilir ve zihinsel sakatlık da ortaya çıkabilir. Leigh sendromuyla ilişkili progresif nörolojik bozulma, genel zayıflık, kas tonusu eksikliği (hipotoni), sakarlık, titreme, kas spazmları (spastisite), bacaklarda yavaş hareketler veya tendon reflekslerinin yokluğudur. Daha fazla nörolojik gelişme gecikir.
Laktik asidoz atakları ortaya çıkabilir ve kanda, beyinde ve vücudun diğer dokularında anormal derecede yüksek laktik asit seviyeleri ile karakterize edilir. Periyodik olarak, kandaki karbondioksit seviyeleri de anormal derecede yükselebilir. Laktik asidoz ve hiperkapni, psikomotor regresyona ve solunum, kalp veya böbrek yetmezliğine yol açabilir.
Leigh sendromlu çocuklar genellikle spontan solunumun geçici olarak durdurulması (apne), solunum zorluğu (dispne), anormal hızlı solunum veya anormal solunum paternleri (Cheyne-Stokes) gibi solunum problemleri geliştirir. Bazı bebekler ayrıca yutma zorluğu yaşayabilir. Görme sorunları arasında anormal derecede hızlı göz hareketleri, halsiz göz bebekleri, şaşılık, bazı göz kaslarının felci, gözlerin sinirlerinin bozulması ve körlüğe yol açan görme bozukluğu olabilir.
Leigh sendromu kalbi de etkileyebilir. Bu bozukluğu olan bazı çocuklar, kalbin anormal genişlemesine ve kalbin çeşitli odalarını bölen fibröz zarın genişlemesine sahip olabilir. Merkezi sinir sistemi dışındaki sinirleri etkileyen hastalıklar ortaya çıkabilir ve bu durum kolların ve bacakların ilerleyici zayıflığına neden olabilir.
Leigh sendromunun X’e bağlı infantil formunun semptomları klasik Leigh sendromununkilere benzer. Erişkinliğin başlangıç formundaki Leigh sendromunun semptomları (subakut nekrotizan ensefalomelopati), hastalığın çok nadir görülen bir şeklidir, genellikle ergenlik döneminde veya erken yetişkinlik döneminde başlar. İlk belirtiler genellikle görme ile ilişkilidir ve bulanık sinirin merkezî görsel alanları (merkezi skotoma), renk körlüğünü veya optik sinirin dejenerasyonundan kaynaklanan progresif görme kaybı gibi anormallikleri içerebilir. Hastalıkla ilişkili nörolojik problemler hastalığın bu formunda yavaş ilerler. Yaklaşık 50 yaşında, etkilenen bireyler, istekli hareketleri koordine etmeyi giderek zor bulabilirler.
Nedenleri
Birçok farklı genetik olarak belirlenmiş metabolik bozukluk tipi Leigh sendromuna neden olabilir. Bu duruma bir veya birkaç farklı enzim eksikliği neden olabilir. Bu enzim eksikliklerine, birkaç farklı hastalık geninden birindeki değişiklikler yani mutasyonlara neden olur. Bu mutasyonlar, otozomal resesif bir özellik, X’e bağlı resesif bir özellik veya mitokondri DNA’sında bulunan bir mutasyon olarak kalıtsal olabilir. Leigh sendromunun bazı vakalarında genetik neden tanımlanamaz. 
Genetik bilgi iki tür DNA’da bulunur: nükleer DNA (nDNA), bir hücrenin çekirdeğinde bulunur ve her iki biyolojik ebeveynden miras alınır. Mitokondriyal DNA (mtDNA), hücrelerin mitokondrilerinde bulunur ve sadece çocuğun annesinden miras alınır. NDNA mutasyonlarına bağlı genetik hastalıklar, biri babadan diğeri de anneden alınan iki gen tarafından belirlenir. Resesif genetik bozukluklar, bir birey her bir ebeveynden aynı özellik için aynı anormal geni aldığında ortaya çıkar. Bir birey hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte yüzde 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi olma riski her hamilelikte yüzde 50’dir. Bir çocuğun her iki ebeveynden normal gen alma ve bu özellik için genetik olarak normal olma şansı yüzde 25’tir.
Diğer DNA bazlı enzim eksiklikleri, bazı otozomal resesif Leigh sendromu vakalarının bir nedeni olarak da gösterilmiştir. Bu spesifik enzim eksiklikleri birkaç farklı genle ilişkilendirilmiştir. Örneğin, kromozom 9 üzerinde bulunan SURF1 geninin mutasyonları sitokrom C oksidaz eksikliğine bağlı Leigh sendromuna neden olur. Bu farklı genetik bozuklukların tümü, merkezi sinir sistemi üzerinde ortak bir etkiye sahip gibi görünmekte ve bu durum ilerleyen nörolojik bozulmalara neden olmaktadır.
Tıbbi literatürde Leigh sendromunun nDNA X’e bağlı resesif formuna dair kanıtlar da vardır. Hastalığın bu formu, X kromozomunun kısa kolunda (p) bulunan piruvat dehidrojenaz kompleksinin E1-alfa alt birimi olarak bilinen bir gendeki spesifik bir kusur ile ilişkilendirilmiştir. X’e bağlı resesif bozukluklar, X kromozomunda kodlanmış durumlardır. Dişiler iki X kromozomuna sahiptir, ancak erkeklerde bir X kromozomu ve bir Y kromozomu vardır. Bu nedenle, kadınlarda, X kromozomundaki hastalık özellikleri diğer X kromozomundaki normal gen tarafından maskelenebilir. Erkeklerde sadece bir X kromozomu bulunduğundan, X’te bulunan bir hastalık için bir gen miras alırlarsa ifade edilir. X’e bağlı bozuklukları olan erkekler, geni taşıyıcıları olan tüm kızlarına, ancak asla oğullarına aktarmaz.
Bazı durumlarda, Leigh sendromu, mitokondri DNA’sında bulunan bir mutasyon olarak anneden kalıtsal olabilir. Vücudun hemen hemen her hücresinde yüzlerce veya binlerce kişi tarafından bulunan Mitokondri, hücresel enerji üretimini düzenler ve bu işlem için genetik planları kendi benzersiz DNA’larında (mtDNA) taşır. Babanın mtDNA’sı sperm hücreleri tarafından taşınır. Bununla birlikte, döllenme sürecinde, babanın mtDNA’sı kaybolur. Sonuç olarak, tüm insan mtDNA’sı anneden gelir. Etkilenen bir anne, özelliklerini tüm çocuklarına geçirecek, ancak yalnızca kızları mutasyonları yeni nesillere geçecektir.
MtDNA’da bulunan genetik mutasyonlar, genlerin normal kopyalarından daha fazla olabilir. Mitokondrinin önemli bir yüzdesinde mutasyonlar mevcut olana kadar semptomlar ortaya çıkmayabilir. Vücudun farklı dokularında normal ve mutant mtDNA’nın eşit olmayan dağılımı aynı aileden bireylerde farklı organ sistemlerini etkileyebilir ve etkilenen aile üyelerinde çeşitli semptomlara neden olabilir.
Leigh sendromunun bazı vakalarından sorumlu olabilecek spesifik mtDNA kusuru ATPase 6 olarak bilinen bir gen ile ilişkilidir. Bu vakalara bazen maternal olarak kalıtsal Leigh sendromu (MILS) veya mtDNA ile ilişkili Leigh sendromu denir.
Bazı araştırmacılar yetişkin başlangıçlı Leigh sendromu vakalarının, bir nDNA mutasyonu nedeniyle otozomal dominant bir özellik olarak kalıtımla alınabileceğine inanmaktadır. Hakim genetik bozukluklar, hastalığın ortaya çıkması için anormal bir genin sadece bir kopyasının gerekli olduğu durumlarda ortaya çıkar. Durum bir nDNA mutasyonuna bağlı olduğundan, anormal gen her iki ebeveynden kalıtsal olabilir veya etkilenen bireyde yeni bir nDNA mutasyonunun sonucu olabilir. Anormal genin etkilenen ebeveynden yavrulara geçme 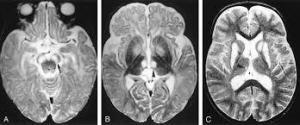 riski, ortaya çıkan çocuğun cinsiyeti ne olursa olsun her hamilelik için yüzde 50’dir.
riski, ortaya çıkan çocuğun cinsiyeti ne olursa olsun her hamilelik için yüzde 50’dir.
Etkilenen Nüfuslar
Leigh sendromunun klasik şekli bebeklik döneminde gelişir ve genellikle 3 ay ile 2 yaş arasında başlar. Bu hastalığın şekli erkekleri ve kadınları eşit sayıda etkiler. Leigh sendromu vakalarında X’e bağlı resesif özellik olarak kalıtsal olanlarda, belirtiler genellikle bebeklik döneminde ortaya çıkmaktadır. Kadınların neredeyse iki katı erkek hastalığının bu şeklinden etkilenir. Bazı nadir durumlarda, Leigh sendromu geç ergenlik döneminde veya erken yetişkinlik döneminde başlayabilir. Kadınların iki katı erkekleri etkileyen bu vakalarda, hastalığın ilerlemesi hastalığın klasik formundan daha yavaştır. Araştırmacılar bir zamanlar klasik Leigh sendromu formunun vakaların yaklaşık yüzde 80’ini oluşturduğuna inanmaktayken tıbbi literatürde Leigh sendromunun prevalansının 36.000-40.000 canlı doğumda 1 olduğu tahmin edilmektedir.
İlgili Bozukluklar
Bozuklukların belirtileri Leigh sendromununkilere benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir. Wernicke sendromu ve Korsakoff sendromu, genellikle tiamin eksikliğinden (B1 vitamini) kaynaklanan hastalıklarla ilişkilidir. Wernicke ensefalopati olarak da bilinen Wernicke sendromu, kafa karışıklığının üçlü triadı, istekli hareketleri ve göz anormalliklerini koordine edememesi ile karakterize nörolojik bir hastalıktır. Korsakoff sendromu, diğer zihinsel yönlerle ilişkili olarak orantısız hafıza kaybıyla karakterize nörolojik bir hastalıktır. Bu iki bozukluk birlikte ortaya çıktığında, Wernicke-Korsakoff sendromu terimi kullanılır. Amerika Birleşik Devletleri’nde, çoğu vaka alkollülerde ortaya çıkmaktadır. Bazı araştırmacılar Wernicke ve Korsakoff sendromlarının ayrı ve ilişkili hastalıklar olduğuna inanmaktayken bazıları aynı bozukluk veya hastalık spektrumunun farklı aşamaları olduğuna inanmaktadır. Wernicke sendromu, daha kısa sürede ve daha ciddi semptomlarla akut faz olarak kabul edilir. Korsakoff sendromu ise kronik olarak kabul edilmektedir ve uzun süren bir sağlık koşuludur.
Nadir bir genetik hastalık olan Batten hastalığı, nöronal seroid lipofusinozları olarak bilinen bir grup ilerleyici dejeneratif nörometabolik hastalığa aittir. Bu bozukluklar benzer semptomları paylaşır ve bu semptomların ortaya çıkma yaşı ile kısmen ayırt edilir. Batten hastalığı, nöronal seroid lipofusinozlarının jüvenil şekli olarak kabul edilir. NCL’ler, beynin sinir hücreleri içindeki bazı yağ, zerre şekilli maddelerin anormal birikmesiyle ve ayrıca ilerleyen bozulmaya karakterize edilir. Beynin belirli bölgelerinde, nörolojik bozukluk, diğer karakteristik belirtiler ve fiziksel bulgulardır. Batten hastalığının semptomları, progresif görme kaybı, nöbetler ve progresif nörolojik dejenerasyon geliştiğinde genellikle 5-15 yaşları arasında belirginleşir. Bazı durumlarda, başlangıç semptomları daha belirsiz olabilir ve sakarlık, denge problemleri ve davranışsal ya da kişilik değişimlerini içerebilir. Batten hastalığı otozomal resesif bir özellik olarak kalıtsaldır ve çoğu Kuzey Avrupa veya İskandinav soyunun ailelerinde görülür.
Tay-Sachs hastalığı, bir enzim eksikliğinin,beyin ve sinir hücrelerinde gangliyozitler olarak bilinen bazı yağların) aşırı birikmesine neden olduğu, nadir görülen, nörodejeneratif bir hastalıktır. Gangliosidlerin anormal olarak birimesi merkezi sinir sisteminde ilerleyen bir işlev bozukluğuna yol açmaktadır. Bu hastalık lizozomal depo hastalığı olarak sınıflandırılır. Lizozomlar hücrelerdeki ana sindirim üniteleridir. Lizozomlardaki enzimler, bazı kompleks karbonhidratlar ve yağlar dahil olmak üzere besinleri parçalamaktadır veya sindirmektedir.
Tay-Sachs hastalığına bağlı semptomlar, ani seslere, abartısızlık, önceden edinilmiş becerilerin kaybı ve aşırı derecede azalmış kas tonusu (hipotoni) için abartılı bir irkilme tepkisi içerebilir. Hastalığın ilerlemesiyle, etkilenen bebekler ve çocuklar gözlerin orta tabakasında kiraz kırmızısı lekeler, kademeli görme kaybı, sağırlık, kas sertliği ve kısıtlı hareketler sonunda felç, beyinde kontrolsüz elektriksel bozukluklar geliştirebilirler. Ve bilişsel süreçlerin bozulması yani demans yaşanabilir. Tay-Sachs hastalığının klasik şekli bebeklik döneminde ortaya çıkar; Ergenlikten 30’lu yaşların ortalarına kadar herhangi bir zamanda yetişkin bir formda ortaya çıkabilir. Tay-Sachs hastalığı, otozomal resesif bir özellik olarak kalıtsaldır.
Nöropati, ataksi ve retinitis pigmentosa (NARP) sendromu nadir görülen bir genetik hastalıktır. Merkezi sinir sistemi dışındaki sinirleri sinir hareketlerini (ataksi) koordine etme kabiliyetini, retinit pigmentosa (RP) olarak bilinen bir göz rahatsızlığını ve çeşitli ek anormallikleri etkileyen sinir hastalıkları ile karakterize edilir. RP, gözlerde astar oluşturan zarın progresif dejenerasyonuna neden olan ve görme bozukluğu ile sonuçlanan bir grup görme bozuklukları için kullanılan genel bir terimdir. Her bir bireydeki NARP sendromunun spesifik semptomları, vakadan duruma büyük ölçüde değişir. Hastalık maternal olarak kalıtsal bir mitokondriyal hastalıktır. NARP sendromuna ATPase 6 geni olarak bilinen mitokondriyal geni etkileyen spesifik bir mutasyon neden olur. Bu mutasyon aynı zamanda maternal kalıtsal Leigh sendromu (MILS) olarak bilinen spesifik bir Leigh sendromu alt türüne de neden olabilir. Aslında, bireyler hücrelerinde mutasyona uğramış mitokondriyal DNA’nın (mtDNA) yüzde 90’ından fazlasına sahip olduğunda, NARP sendromu değil MILS olarak sınıflandırılırlar. NARP sendromlu çoğu birey mutasyona uğramış mtDNA’nın yüzde 70-80’ine sahiptir.
Teşhis
Leigh sendromu tanısı kapsamlı bir klinik değerlendirme ve özellikle ileri görüntüleme teknikleri gibi çeşitli uzmanlık testleri ile doğrulanabilir. Beynin manyetik rezonans  görüntüleme (MRG) veya bilgisayarlı tomografi (BT) taramaları, beynin belirli bölgelerinde anormal alanları ortaya çıkarabilir. Bir MRG, belirli organların ve vücut dokularının enine kesit görüntülerini üretmek için bir manyetik alan ve radyo dalgaları kullanır. BT taraması sırasında, bir bilgisayar ve röntgenler, belirli doku yapılarının kesit görüntülerini gösteren bir film oluşturmak için kullanılır.
görüntüleme (MRG) veya bilgisayarlı tomografi (BT) taramaları, beynin belirli bölgelerinde anormal alanları ortaya çıkarabilir. Bir MRG, belirli organların ve vücut dokularının enine kesit görüntülerini üretmek için bir manyetik alan ve radyo dalgaları kullanır. BT taraması sırasında, bir bilgisayar ve röntgenler, belirli doku yapılarının kesit görüntülerini gösteren bir film oluşturmak için kullanılır.
Beynin beyin korteksinde küçük veya büyük kistler bulunabilir. Laboratuar testleri, kanda (laktik asidoz) yüksek seviyelerde asidik atık ürünlerin yanı sıra yüksek seviyede piruvat ve alanin ortaya çıkarabilir. Kan şekeri (glukoz) normalden biraz daha düşük olabilir. Enzim piruvat karboksilaz karaciğerden bulunmayabilir ve etkilenen kişilerin kanında ve idrarında bir tiamin trifosfat (TTP) üretimi inhibitörü bulunabilir. Leigh sendromlu bazı çocuklar, enzimlerin piruvat dehidrojenaz kompleksi veya sitokrom C oksidazın tespit edilebilir eksikliklerine sahip olabilir.
Tedavi
Herhangi bir tür Leigh Sendromu için kanıtlanmış bir tedavi yoktur. Tedavi önerileri temel olarak vaka raporları ve kişisel gözlemlere dayanmaktadır. Leigh sendromunun tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını gerektirebilir. Çocuk doktorları, kardiyologlar, nörologlar, işitme problemlerini değerlendiren ve tedavi eden uzmanlar göz uzmanları ve diğer sağlık profesyonellerinin etkili bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilir.
Leigh sendromu için en yaygın tedavi tiamin (Vitamin B1) veya tiamin türevlerinin verilmesidir. Bu bozukluğu olan bazı kişiler geçici semptomatik iyileşme ve hastalığın ilerlemesinde hafif bir yavaşlama yaşayabilir. Piruvat dehidrojenaz enzim kompleksi eksikliği de olan Leigh sendromlu hastalarda yüksek yağ oranı düşük karbonhidrat diyeti önerilebilir. Görme engelli ikişilere fayda sağlayan hizmetler, Leigh sendromlu bazı kişiler için de rahatsızlığın giderilmesinde yardımcı olabilir. Bu bozukluğu olan etkilenen bireylerin ailelerine genetik danışmanlık önerilmektedir. Diğer tedavi semptomatik ve destekleyicidir.
Yazar: Özlem Güvenç Ağaoğlu