
 Prion; insanlarda Creutzfeldt-Jakob hastalığı (CJD) da dahil memelilerde birkaç farklı nörodejeneratif hastalıktan sorumlu enfekte edici bir ajandır. Enjeksiyona neden olan bu ajan sadece proteinden oluşur ve nükleik asit yapısı (DNA ya da RNA molekülü) içermez. Bu yönleriyle çoğalmalarını sağlayan nükleik asitleri içeren virüslerden ve bakterilerden oldukça farklıdırlar. Bu yapı da prionların nükleik asitleri parçalayan ultraviyole ışınlara nasıl dirençli olduğunu açıklar. Fakat ultraviyole ışınların protein yapılara da zarar verdiğinden şüphelenilir.
Prion; insanlarda Creutzfeldt-Jakob hastalığı (CJD) da dahil memelilerde birkaç farklı nörodejeneratif hastalıktan sorumlu enfekte edici bir ajandır. Enjeksiyona neden olan bu ajan sadece proteinden oluşur ve nükleik asit yapısı (DNA ya da RNA molekülü) içermez. Bu yönleriyle çoğalmalarını sağlayan nükleik asitleri içeren virüslerden ve bakterilerden oldukça farklıdırlar. Bu yapı da prionların nükleik asitleri parçalayan ultraviyole ışınlara nasıl dirençli olduğunu açıklar. Fakat ultraviyole ışınların protein yapılara da zarar verdiğinden şüphelenilir.
Prionun keşfi, bilim insanlarının normal bir hücrenin zarında anormal şekilli bir protein bulduklarında gerçekleşti. Bazı bilim insanları da yapısı bozuk proteinin aynı türde başka proteinlere bağlandığını ve bağlandıkları proteinlerin şeklini değiştirdiğini düşündüler. Böylece hastalığın yayılması için bir zincirleme reaksiyon ortaya çıkar ve yeni enfekte edici materyaller üretilir. Bu proteini kodlayan gen başarılı bir şekilde klonlandı ve fare modelinin kullanıldığı çalışmalar prion hipotezini doğruladı. İnkar edilemez olmasalar da, şu an bu hipotezi destekleyen bilimsel kanıtlar oldukça sağlamdır.
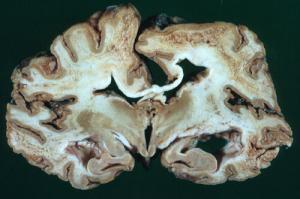 Prion hastalıklarına yönelik araştırmalar son yıllarda artmıştır. Bu artışın birincil sebebi ise, prionların çok sayıda büyükbaş hayvanı etkileyen deli dana hastalığına neden olması ve bu hayvanların ürünlerinin tüketilmesiyle ölümcül olan bu hastalığın insanlara da bulaşmasıdır. Deli dana hastalığı, dünya çapında ciddi bir sağlık sorunu olarak kendini göstermiştir.
Prion hastalıklarına yönelik araştırmalar son yıllarda artmıştır. Bu artışın birincil sebebi ise, prionların çok sayıda büyükbaş hayvanı etkileyen deli dana hastalığına neden olması ve bu hayvanların ürünlerinin tüketilmesiyle ölümcül olan bu hastalığın insanlara da bulaşmasıdır. Deli dana hastalığı, dünya çapında ciddi bir sağlık sorunu olarak kendini göstermiştir.
Prion terimi ilk defa 1982’de Kaliforniya Üniversitesi’nden Stanley B. Prusiner tarafından koyunlarda görülen bir hastalığı tanımlamak için kullanıldı. Bu aşamadan sonra, hastalığın bir virüs ya da bakteri yerine anormal yapıda bir protein tarafından ortaya çıktığı görüldü.
Proteinin yapısındaki anormal değişime neden olan süreç henüz kesin değildir ve normal ve anormal formda olan prion proteinin yapısını belirlemeye yönelik çok sayıda çalışma vardır. Yakın zamanda bilim insanları her iki form içinde bir moleküler model geliştirdiler ve prion proteinin yapısını tanımlayan bir makale yayınladılar. Manyetik rezonans görüntüleme ve X ışını kristalografisi kullanılan çalışmalar prionun temel yapısını anlamada bize yardımcı olur.
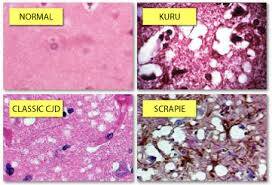 Creutzfeldt-Jakob hastalığı (CJD), kuru and Gerstmann-Str¿ussler-Scheinker (GSS) hastalığı gibi ölümcül ve insanlarda görülen nörodejeneratif hastalıkların nedeninin prionlar olduğu düşünülür.
Creutzfeldt-Jakob hastalığı (CJD), kuru and Gerstmann-Str¿ussler-Scheinker (GSS) hastalığı gibi ölümcül ve insanlarda görülen nörodejeneratif hastalıkların nedeninin prionlar olduğu düşünülür.
U.S. National Institutes of Health’de çalışan Dr. Carleton Gajdusek, kuru ya da CDJ’den ölen insanların beyninlerinden hazırladıkları ekstraktları şempanzelerin beynine aşıladıklarında onlarda da hastalığın meydana geldiğini gördüler. Bu deneyler enfekte edici bir ajanın varlığını göstermiştir. Bu çıkarım, CDJ’lerin hastalara kornea nakli ve büyüme hormonu terapisi gibi çeşitli tıbbi tedaviler sırasında kazayla bulaşmasıyla doğrulandı.
Kafa karıştırıcı şekilde GSS gibi bazı prion hastalıkları da kalıtımsaldır. Kalıtım şekli, otozomal ve dominant olarak tanımlandı. Bunun anlamı; eğer ebeveynlerden birinde GSS varsa, herhangi bir cinsiyetten olan çocuğunda bu hastalığın görülmesi olasılığı %50’dir.
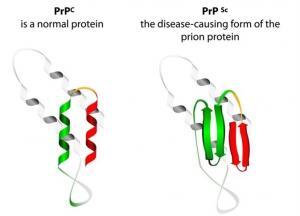
Prionların hastalık yapıcı mekanizmasıyla ilgili evrensel olarak kabul edilmiş bir açıklama yoktur fakat bu konudaki çalışmalar devam etmektir. Prionun normal koşullarda PrP isimli bir protein olarak kodlandığını biliyoruz ama normal koşullardaki PrP’nin yapısı bozulduğunda priona ve hastalıklara neden olur. Normal yapıdaki PrP proteini bu protein hepimizde bulunur ve 250 amino asitten oluşur. PrP proteinini kodlayan genin nöronları koruma ve nöronlar arasında iletişimi sağlama görevleri vardır.
 Bazı bilim insanları da PrP’nin yabancı patolojik nükleik asitlerle temas ettiği zaman prion formuna dönüştüğünü iddia ediyorlar. Bu iddia virino hipotezi olarak adlandırılır. Virino hipotezini destekleyen bulgu, farklı prion tiplerinin varlığıdır. Fakat, bilim insanları prionlarla ilişkili herhangi bir nükleik asit bulamamışlardır. Ayrıca, dokudaki nükleik asitlerin parçalanmasının ardından prionlar enfekte edici olmayı sürdürüyorlardı.
Bazı bilim insanları da PrP’nin yabancı patolojik nükleik asitlerle temas ettiği zaman prion formuna dönüştüğünü iddia ediyorlar. Bu iddia virino hipotezi olarak adlandırılır. Virino hipotezini destekleyen bulgu, farklı prion tiplerinin varlığıdır. Fakat, bilim insanları prionlarla ilişkili herhangi bir nükleik asit bulamamışlardır. Ayrıca, dokudaki nükleik asitlerin parçalanmasının ardından prionlar enfekte edici olmayı sürdürüyorlardı.
Şu an yaygın şekilde kabul gören teori ise prionun herhangi bir nükleik asitle ilişkili olmayan tek başına bir ajan olmasıdır. PrP hücrelerde normalde bir hastalığa yol açmayacak şekilde sentezlenir. Bu proteinin yapısı bozulabilir ve hastalığa neden olan anormal bir şekle girebilir. Çünkü anormal şekil, enfeksiyon yapıcı özellik taşır ve normal formdaki proteinle de etkileşime girerek onun da anormal forma geçmesine neden olur. Böylece dokunun içerisinde anormal yapıdaki protein miktarı sürekli artar.
Prionlar bulaşıcıdır ve yemeyle ya da aşılamayla bulaşabilir. Ayrıca ileri yaşlarda prion hastalığının kendiliğinden ortaya çıkma ihtimali vardır. Buna ek olarak, kalıtsal CJD ve GSS hastalıkları PrP genindeki mutasyonlardan (DNA dizisindeki değişim) kaynaklı olabilir. Böylece PrP proteinin amino asit (proteinlerin yapıtaşı olan moleküller) dizisi değişir ve sonuç olarak yapısı da değişir.
PrP’nin yapsının fiziksel analizi iki farklı proteinin (normal yapıdaki ve hastalık yapıcı anormal yapıdaki) varlığını doğrular. Bu analiz, manyetik rezonans görüntüleme analizi ile yapılmıştır.
Prionların beyinde nörodejeneratif hastalıklara nasıl neden olduğunun mekanizması henüz bilinmiyor. Fakat, beyin dokusunda biriktiklerini ve hücrelerin ölümüne neden olduğunu biliyoruz. Dünya çapında çok sayıda büyükbaş hayvanın ölümüne neden olan ve insan sağlığını tehdit eden deli dana hastalığından iyi hijyen uygulamalarıyla korunmak mümkündür. Fakat deli dana hastalığı dışında da var olan kalıtımsal prion kaynaklı hastalıklardan daha etkili şekilde korunabilmek ve tedavi stratejileri geliştirebilmek için prionlarla ilgili moleküler mekanizmalara ilgili daha fazla bilimsel çalışma yapılmasına ihtiyaç vardır.
Kaynakça:
https://ghr.nlm.nih.gov/condition/prion-disease#genes
https://www.scientificamerican.com/article/what-is-a-prion-specifica/
Yazar: Ayça Olcay